Visualizing regulated necroptosis execution in live cells has been possible through inducible expression of a minimal truncated MLKL construct, NBB140-2xFV-Venus. This construct maintains the ability to induce plasma membrane permeabilization and is activated through Dim-induced oligomerization of the FKBP cassette (2xFV). We observe and quantify necroptosis by live-cell microscopy imaging, monitoring kinetically (every 5 min) the uptake of a cell impermeable green fluorescence DNA binding dye (Figure 6A). This inducible system is very robust, and complete necroptosis of mlkl-/- MEFs can be observed within ~1 h upon Dim-mediated oligomerization of Dox-preincubated cells that express NBB140-2xFV-Venus (Figure 1A). We refer to these conditions as fast-kinetics necroptosis. Expression of Venus alone ± Dim or NBB140-2xFV-Venus in the absence of Dim did not induce necroptosis (Figure 6A). We usually perform at least 3 replicate imaging experiments in triplicate or quadruplicate in 24-, 96-, or 384-well plates.
The fast-kinetics necroptosis induced by enforced dimerization of NBB140-2xFV-Venus (Figure 6A) supports the role of MLKL as the putative executioner of plasma membrane rupture. To visualize the redistribution of NBB140-2xFV-Venus to the plasma membrane during necroptosis, live-cell confocal microscopy is used to monitor Venus fluorescence. During fast-kinetics necroptosis, within 2–3 min of incubation with Dim, Venus coating of the cell periphery is observed, followed by gradual cell rounding (Movie 1). NBB140-2xFV-Venus accumulation at the plasma membrane is visualized by TIRF confocal microscopy, which focuses on the cell volume at the cytosol-plasma membrane-glass interface. Plasma membrane-associated Venus puncta are visible within 1–2 min of incubation with Dim (Movie 2). Thus, enforced oligomerization of NBB140-2xFV-Venus induces its rapid redistribution to plasma membrane.
Scanning electron microscopy (SEM) is a powerful tool to reveal the morphological changes in cells undergoing necroptosis. Under fast necroptosis induced by oligomerization of NBB140-2xFV-Venus, cells change morphology from normal elongated shapes (time 0 min) to rounded and swelled (5 min) to partially ruptured (10 min) and extensively dismantled where the cytosol has vanished (20 min) (Figure 6B). SEM complements the previous observations from live-cell microscopy correlating MLKL localization to the plasma membrane with membrane rupture. Overall the microscopy techniques presented herein offer complementary means to monitor, evaluate and quantify necroptosis at cellular level and implicate NBB140-2xFV-Venus in the execution of plasma membrane rupture.
To determine if MLKL can directly contact the plasma membrane we perform in vitro lipid binding experiments monitored by NMR spectroscopy using recombinant NBB156. Serial dilutions of lipid-detergent micelles, using a constant concentration of detergent (vehicle for lipid presentation) and variable lipid concentrations, are tested for binding to 15N-labeled NBB156 by 2D NMR spectroscopy, which monitors binding-induced changes in the protein. NBB156 undergoes major structural changes upon lipid binding to displace the inhibitory brace region (amino acids 132-156) from the closed and helical (Closed Brace) to the open and intrinsically disordered conformation (Open Brace) (Figure 7A). Two-dimensional NMR spectroscopy provides per residue information on mixtures of both conformers (Figures 7B-7C). We previously assigned the backbone amides of both conformers24. We can easily monitor the percentages of closed and open conformers in a given sample as described in Figures 5A-C. Using this binding assay, we explore NBB156 binding to PIPs in DDM detergent, which is inert to NBB156 binding (Figure 5B). In this assay, PIP2 is the best NBB156 ligand, which induces full opening of the inhibitory brace at 125 µM (Figure 5C). In contrast, saturated (18:0) PI and unsaturated (18:1) PI are poor NBB156 ligands inducing partial brace opening under the same conditions (Figure 5C). Our NMR-based lipid binding assay provides supporting evidence for the interaction of MLKL NBB156 with phospholipids and suggests a direct link between MLKL and the plasma membrane.
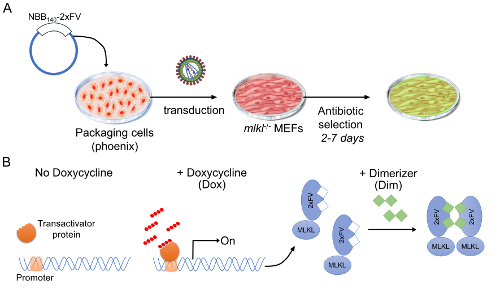
Figure 1: Stable cell line harboring a modified Tet-On 3G inducible system. (A) Stable cell lines were generated by retroviral transduction of mlkl-/- MEFs with the pTREX-rtTA-blast followed by the pRetroX-TRE3G-NBB140-2xFV-Venus-puro. Each transduction was followed by antibiotic selection for up to 1 week before moving on to subsequent analyses. (B) Schematic representation of inducible MLKL expression system. This system is based on 2 drug-inducible regulatory steps: i) MLKL gene expression and protein production (+Dox) and ii) activation by oligomerization (+Dim). When protein production is induced in advance, +Dox, fast-kinetics necroptosis can be triggered, +Dim. Please click here to view a larger version of this figure.
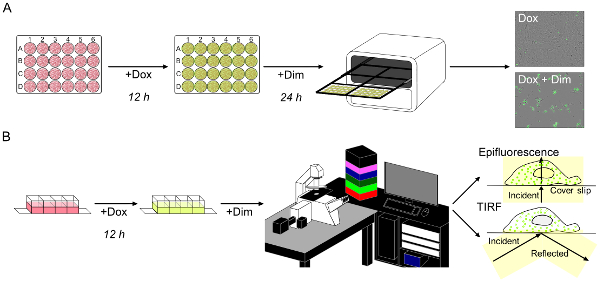
Figure 2: Live-cell imaging of fast-kinetics necroptosis reveals rapid membrane relocalization of MLKL. (A) Fast-kinetics necroptosis induced as in Figure 1B can be monitored in an imaging system. Necroptosis is scored by uptake of the cell-impermeable green fluorescent dye. (B) Schematic representation of live-cell imaging of fast-kinetics necroptosis with epifluorescence and total internal reflection fluorescence (TIRF) microscopy analysis. Please click here to view a larger version of this figure.
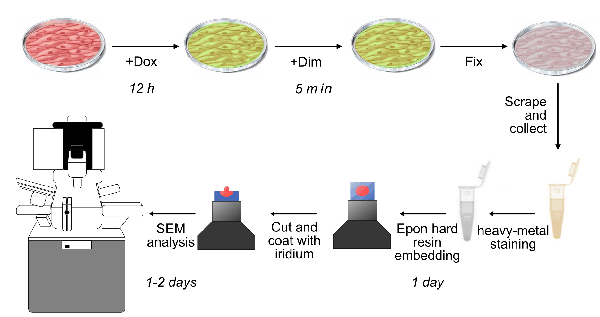
Figure 3: Schematic representation of scanning electron microscopy (SEM) sample preparation and analysis. Cells from fast-kinetics necroptosis induced as described in Figure 1 are fixed on the plate at different time points after addition of Dim. The cells are then prepared for SEM analysis by scraping and pooling, heavy metal staining, resin embedding, and iridium coating as described in section 4. Please click here to view a larger version of this figure.
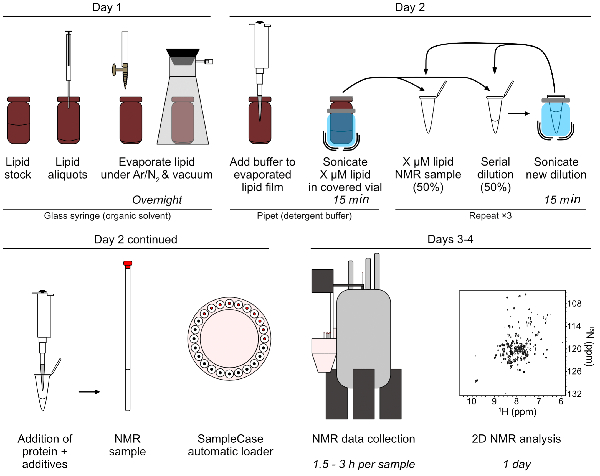
Figure 4: Sample preparation and data collection for NMR titrations of 15N NBB156 and lipid-detergent micelles. This assay and analysis is typically performed in 3–4 days: During day 1, lipid aliquots are dispensed and dried overnight. During day 2, the lipid films are rehydrated in assay buffer ± detergents, sonicated, and serially diluted (two-fold) by mixing equal volumes of the rehydrated lipid solution with lipid-free buffer solution. Each dilution is sonicated to ensure homogeneous mixing and distribution of lipids in detergent micelles prior to subsequent dilutions. Protein samples are mixed in the serial lipid-detergent micelle dilutions, loaded in the appropriate NMR tubes, placed in a SampleCase loader (or loaded manually), and automatic NMR data collection is started. During subsequent days, NMR data collection is completed and followed by 2D NMR analysis. Please click here to view a larger version of this figure.
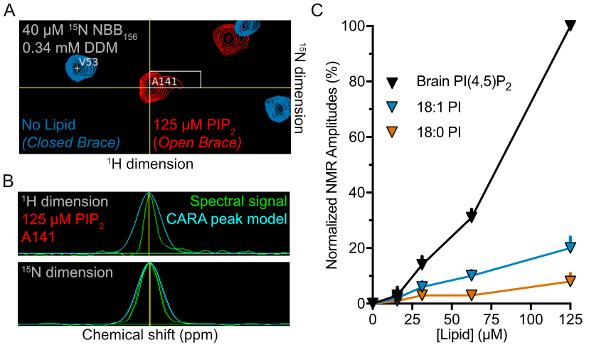
Figure 5: Lipid-binding preferences of NBB156 measured using 2D NMR. (A) Superimposed 1H-15N TROSY spectra for 15N-NBB156 in the presence (red) or absence (blue) of 125 µM PI(4,5)P2 in 0.34 mM DDM visualized in CARA. (B) One-dimensional slices of resonances used for amplitude measurements and normalization. The raw spectrum (green) is overlaid with the boundary for amplitude and integration by the program CARA. (C) Normalized amplitudes of NBB156 in the presence of phosphoinositide-DDM micelles plotted against lipid concentration. PIP2 induces full opening of the brace. Please click here to view a larger version of this figure.
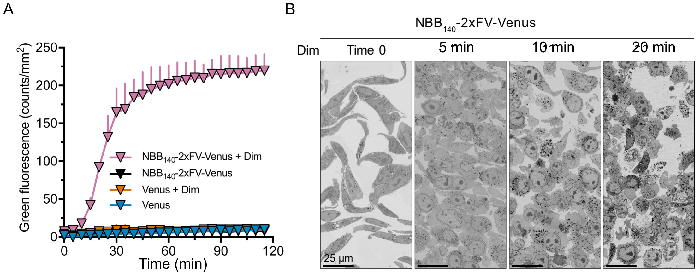
Figure 6: Fast-kinetics necroptosis induced by oligomerization of NBB140-2xFV-Venus in mlkl-/- MEFs. (A) Necroptosis quantification by high-throughput fluorescence imaging of uptake of cell-impermeable, DNA-binding green fluorescent dye. Robust necroptosis induction is only observed upon Dim-induced oligomerization of NBB140-2xFV-Venus, but not in the absence of Dim. Venus-only control experiments are also performed. All conditions are done in triplicate and are plotted as average and SD from one representative replicate. (B) Fast-kinetics necroptosis induced in the presence of Dim visualized with scanning electron microscopy. The time course reveals morphological changes underlying necroptosis including cell rounding and swelling (5 min), rupture of plasma membrane (10 min), and complete extravasation of the cytosol (20 min). Each sample had similar number of cells at time 0 min. From left to right, the zoomed in area contains 26, 32, 23, and 36 cells with nuclei. Please click here to view a larger version of this figure.
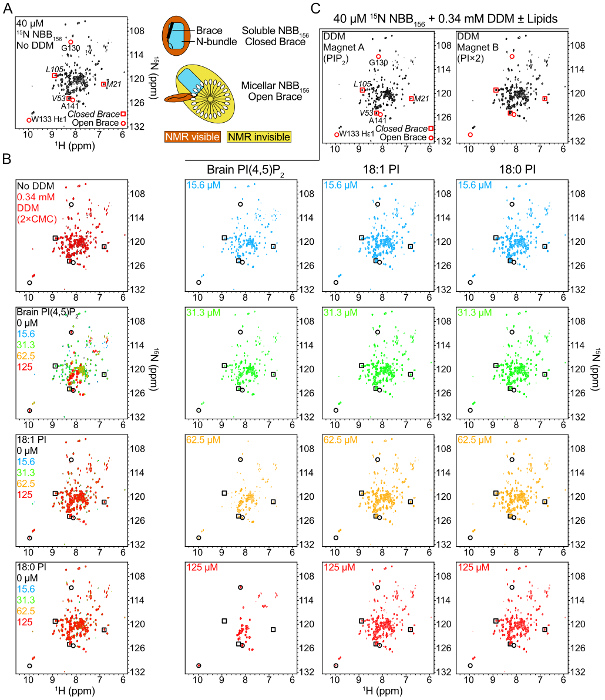
Figure 7: Binding of NBB156 to phosphoinositides monitored by NMR spectroscopy. (A) 1H-15N TROSY spectrum of 15N-NBB156. The chemical shifts of resonances used for normalization of the closed brace spectrum or quantification of the open state are highlighted with squares or circles, respectively. Right, schematic of NMR signatures detected in the absence or presence of lipid-detergent micelles. NBB156 does not bind DDM detergent micelles in the absence of phosphoinositides. (B) Superimposed 1H-15N TROSY spectra of 15N-NBB156 in the presence of the respective lipid-detergent micelles. (C) Single 1H-15N TROSY spectra of 15N-NBB156 in the presence of the respective lipid-detergent micelles from panel B. By detecting the open and closed conformations unambiguously in mixtures of the two conformations we can quantify the percentages of the two conformers in a given sample. PIP2 is the preferred lipid ligand of NBB156 providing a direct link between MLKL and plasma membrane phospholipids. Please click here to view a larger version of this figure.
| PCR buffer (10x) | 5.0 µL |
| DNA template (100 ng/µL) | 1.0 µL |
| cDNAs for MLKL, 2x FKBP, Venus) | |
| dNTPs (25 mM each NTP) | 0.5 µL |
| PCR primers forward (F) (100 ng/µL) | 1.3 µL |
| PCR primers forward (FR (100 ng/µL) | 1.3 µL |
| NBB140 F | ATAATCGATACCATGGAAAATTTGAAGCATATT |
| NBB140 R | TATGCGGCCGCATCCTGCTGATCTTCCTGTGC |
| 2xFKBP F | ATAGCGGCCGCAGGCGTCCAAGTCGAAACCATT |
| 2xFKBP R | TATGCGGCCGCTTCCAGTTTTAGAAGCTCCAC |
| Venus F | ATAGGGCCCACCATGGTGAGCAAGGGCGAG |
| Venus R | TATGAATTCTTACTTGTACAGCTCGTC |
| DNA Polymerase (2.5 U/µL) | 1.0 µL |
| Distilled deionized water (ddH2O) | 39.9 µL |
| Total reaction volume | 50.0 µL |
| PCR cycling parameters | |
| 1 cycle | 94-98 °C; 45 s |
| 25–30 cycles | 94-98 °C; 45 s |
| 58 °C; 45 s | |
| 72 °C; 1-2 min | |
| 1 cycle | 72 °C; 10 min |
Table 1: PCR reaction of NB140-2xFV-Venus for restriction enzyme-based cloning in pRetroX-TRE3G.
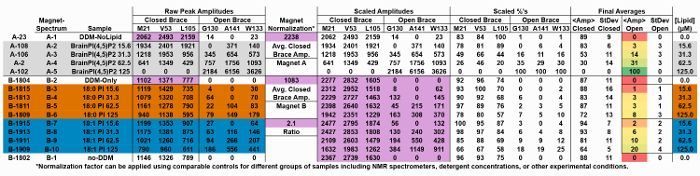
Table 2: Raw data for NMR spectra presented in Figure 5 illustrating the normalized transformation of data collected from a second NMR magnet to the calculated average open brace state. Please click here to view a larger version of this table.
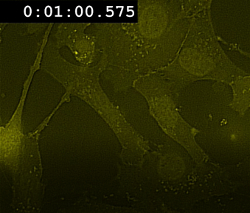
Movie 1: Fast-kinetics necroptosis induced by oligomerization of NBB140-2xFV-Venus in mlkl-/- MEFs. Live confocal microscopy showing the rapid translocation of NBB140-2xFV-Venus from the cytosol to the plasma membrane after enforced oligomerization of the FKBP domain. The cells were treated as described in Figure 1. Yellow: NBB140-2xFV-Venus. Please click here to view this video. (Right-click to download.)
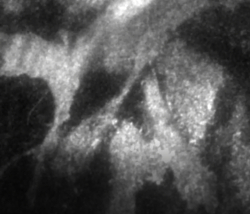
Movie 2: TIRF microscopy of fast-kinetics necroptosis. TIRF microscopy showing fast (~2 min) relocalization and aggregation of MLKL on the plasma membrane upon addition of Dim to mlkl-/- MEFs expressing NBB140-2xFV-Venus. Please click here to view this video. (Right-click to download.)
