Sensitive Measurement of Mitophagy by Flow Cytometry Using the pH-dependent Fluorescent Reporter mt-Keima
Summary
Mitophagy, the selective degradation of mitochondria, has been implicated in mitochondrial homeostasis and is deregulated in various human diseases. However, convenient experimental methods for measuring mitophagy activity are very limited. Here, we provide a sensitive assay for measuring mitophagy activity using flow cytometry.
Abstract
Mitophagy is a process of selective removal of damaged or unnecessary mitochondria using autophagy machinery. Close links have been found between defective mitophagy and various human diseases, including neurodegenerative diseases, cancer, and metabolic diseases. In addition, recent studies have shown that mitophagy is involved in normal cellular processes, such as differentiation and development. However, the precise role of and molecular mechanisms underlying mitophagy require further study. Therefore, it is critical to develop a robust and convenient method for measuring changes in mitophagy activity. Here, we describe a detailed protocol for quantitatively assessing mitophagy activity through flow cytometry using the mitochondria-targeted fluorescent protein Keima (mt-Keima). This flow cytometry assay can analyze mitophagy activity more rapidly and sensitively than conventional microscopy- or immunoblotting-based methods. This protocol can be applied to analyze mitophagy activity in various cell types.
Introduction
Mitochondria are organelles that are essential for cell proliferation and physiology. Mitochondria are responsible for generating more than 80% of the ATP supply via oxidative phosphorylation, and they also provide various metabolic intermediates for biosynthesis and metabolism1,2. In addition to their roles in energy supply and metabolism, mitochondria play central roles in many other important processes, including reactive oxygen species (ROS) generation, the regulation of cell death, and intracellular Ca2+ dynamics3. Alterations in mitochondrial function have been associated with human diseases, including cancer, diabetes mellitus and various neurodegenerative diseases4,5. Increased mitochondrial dysfunction and mitochondrial DNA mutations are also thought to contribute to normal aging processes6,7,8. Therefore, quality control is a crucial issue in mitochondria. Mitochondria are highly dynamic organelles that can continuously change their shape between elongated networks and short, fragmented form. Mitochondria dynamics plays an important role in maintaining the function of mitochondria as well as their degradation through mitophagy9.
Mitophagy is a mechanism that involves the selective degradation of whole mitochondria using the autophagy machinery. Mitophagy is the principle mechanism underlying mitochondrial turnover and the removal of damaged or dysfunctional mitochondria. In this process, mitochondria are first surrounded by a membrane, resulting in the formation of autophagosomes, which then fuse with lysosomes for hydrolytic degradation9. Previous genetic studies in Drosophila have suggested that two Parkinson's disease-related genes, PTEN-induced putative kinase 1 (PINK1) (PARK6) and Parkin (PARK2), function in the same pathway10,11. Subsequence studies have shown that the PINK1-Parkin pathway is responsible for eliminating damaged mitochondria and defects in this pathway result in dysfunctional mitophagy and may contribute to human diseases12,13. Defects in mitophagy processes have recently been found in various human diseases, including cancer, heart disease, liver diseases and neurodegenerative disease 14. In addition, recent studies have also shown that mitophagy is critical to many physiological processes, such as differentiation, development, and the immune response15,16,17,18,19, suggesting that mitophagy may play a more active role in controlling cell functions.
Despite recent confirmation that mitophagy plays an important role in both quality control in mitochondria and human disease, the molecular mechanisms underlying mitophagy remain poorly understood. Although mitophagy-related mechanisms have been systematically studied in yeast, studies aimed at exploring mitophagy-related mechanisms in mammalian cells have mainly focused on the PINK1-Parkin pathway. Previous studies have established that the PINK1-Parkin pathway is primarily responsible for the selective removal of damaged mitochondria via mitophagy12,20,21. However, recent studies have reported that in some models, mitophagy can be activated even in the absence of functional PINK122,23,24. These results suggest that in addition to the PINK1-Parkin pathway, there an additional unidentified pathway that can activate mitophagy.
The absence of a convenient method to assess mitophagy activity is a major obstacle to exploring the pathways that regulate mitophagy and its role in pathophysiological events. Electron microscopy is a powerful tool to directly visualize autophagosome-engulfed mitochondria. However, electron microscopy has limitations in quantifying mitophagy activity. Although strategies that use microtubule-associated protein-1 light chain 3 (LC3)-conjugated fluorescent probes, such as GFP-LC3, are currently the most widely used approaches25, the transient nature of the LC3-based signal and its high rate of false-positive signals limit its sensitivity for detecting mitophagy in cells26. A combination of western blotting to measure mitochondrial protein level, the quantification of mitochondrial DNA, and fluorescence microscopy analysis to colocalize mitochondria with either autophagosomes or lysosomes would be a good approach for assessing mitophagy. However, quantification limitations and a lack of convenience of existing methodologies call for new approaches. The introduction of a new pH-dependent fluorescent protein, the mitochondrial target Keima (mt-Keima), greatly improved the ability to detect mitophagy27. By fusing the mitochondrial-targeting sequence of cytochrome c oxidase subunit VIII (COX VIII) into Keima, mt-Keima is directed to the mitochondrial matrix. The large shift in the excitation peak of Keima from 440 nm to 586 nm (corresponding to pH 7 and pH 4, respectively) can be utilized to assess mitophagy with good sensitively in both in vitro and in vivo experiments28,29. More importantly, the resistance of mt-Keima to lysosomal degradation causes the integration of the mt-Keima signal in lysosomes, allowing for the easy quantitative measurement of mitophagy activity. The fluorescence change that occurs in mt-Keima can be analyzed using either confocal microscopy or flow cytometry28,29. However, a flow cytometry-based method for measuring mitophagy activity using mt-Keima has not been provided in detail to date.
Here, we describe a detailed protocol for a flow cytometry-based technique for measuring mitophagy activity in cells using mt-Keima. Although we have shown here that flow cytometry analysis successfully detected CCCP-induced mitophagy in HeLa cells expressing Parkin, this technique can be applied to a wide variety of cell types.
Protocol
1. Generation of HeLa cells expressing mito-Keima (mt-Keima)
- Preparation of mt-Keima lentivirus
- Coat a 100-mm culture dish by adding 2 mL of 0.001% poly-L-lysine/phosphate-buffered saline (PBS) and allow it to stand for 5 min at room temperature.
- Remove the poly-L-lysine solution using a glass pipette connected to a vacuum and wash the culture dish by adding 2 mL of 1x PBS.
- Plate 1.5 x 106 HEK293T cells on the coated culture dish with 10 mL of DMEM containing 10% FBS and 1% penicillin/streptomycin, and culture the cells at 37 °C in a CO2 tissue culture incubator for one day.
- Transfect 2 µg of mt-Keima lentiviral plasmid29 DNA together with packaging DNAs (psPAX 2 µg and pVSVG 0.25 µg) using a transfection reagent for 8 h according to the manufacturer's instructions.
- Remove the transfection media and add 8 mL of fresh media.
- Collect the media containing viral particles 48 h later. Remove cellular debris from the collected media by centrifugation at 500 x g for 5 min and filter them with a 0.45-µm syringe filter.
Note: For long-term use, make aliquots of lentiviral particles and store the samples at -80 °C. Avoid subjecting the viral samples to freeze-thaw cycles for efficient viral infection.
- Infecting HeLa-Parkin cells with mt-Keima lentivirus
- Plate 5 x 104 HeLa cells expressing Parkin (HeLa-Parkin) in a 60-mm culture dish with 4 mL of DMEM containing 10% FBS and 1% penicillin/streptomycin at 37 °C one day before harvesting the mt-Keima lentivirus.
Note: Because of the absence of endogenous Parkin expression in HeLa cells30, the HeLa-Parkin cells used here to more clearly show CCCP-induced mitophagy. This procedure can also be applied to other primary or immortalized cell lines. - Remove the growth medium the next day and add 1 mL of mt-Keima lentiviral media. Add an additional 2 mL of growth medium containing 3 µL of polybrene stock solution (8 mg/mL in distilled water). Incubate for one day in a CO2 tissue culture incubator.
- Remove the mt-Keima lentiviral mixture the next day and wash the cells twice with 1x PBS. Add 4 mL of growth medium and treat the cells with 2 µg/mL puromycin for two days.
- After two days of puromycin selection, remove the growth medium, and wash the cells twice with 1x PBS. Add growth medium, and culture the cells in a CO2 incubator until use.
Note: This method can be applied to generate cells that stably express mt-Keima, including other cell lines and primary cells.
- Plate 5 x 104 HeLa cells expressing Parkin (HeLa-Parkin) in a 60-mm culture dish with 4 mL of DMEM containing 10% FBS and 1% penicillin/streptomycin at 37 °C one day before harvesting the mt-Keima lentivirus.
2. Inducing mitophagy using CCCP treatment and preparing FACS samples
- Plate 5 x 104 HeLa-Parkin cells expressing mt-Keima in a 60-mm culture dish with 4 mL of DMEM containing 10% FBS and 1% penicillin/streptomycin and culture them for one day at 37 °C in a CO2 tissue culture incubator.
- Remove the growth medium the next day and add 4 mL of fresh DMEM containing 10 µM carbonyl cyanide m-chlorophenylhydrazone (CCCP). Incubate HeLa-Parkin-mt-Keima cells in a CO2 tissue incubator for 6 h.
- Remove the growth medium 6 h later and wash the cells with 2 mL of 1x PBS. Detach the cells by treating them with trypsin/EDTA solution and inactivate the trypsin by adding growth medium containing 10% FBS. Transfer the cells into a 15-mL conical tube.
- Centrifuge the cells at 500 x g for 5 min.
- Carefully remove the growth medium by aspiration and resuspend the cells with 1 mL of cold 1x PBS.
- Transfer cells into the appropriate FACS tube and place it on ice.
Note: Prepare uninfected HeLa cells as a negative control.
Note: Cells are viable on ice for several hours, and mt-Keima fluorescence signals also remain stable.
3. FACS analysis of mitophagy
Note: In this study, cells were analyzed with a flow cytometer equipped with a 405-nm and 561-nm laser. Cells were excited with a violet laser (405 nm) with emission detected at 610 ± 10 nm with a BV605 detector and with a yellow-green laser (561 nm) with emission detected at 610 ± 10 nm by a PE-CF594 detector simultaneously (Figure 1).
- Gating live cells
- Turn on the flow cytometer and computer and log into the analysis software.
- Generate a new experiment or duplicate an existing experiment. To excite cells with the violet (405 nm) and yellow-green (561 nm) lasers and detect emission at 610 ± 10 nm detector, select BV605 and PE-CF594 in addition to forward scatter (FSC) and side scatter (SSC) in the parameter window as required fluorophores.
Note: All fluorophores should be added in a linear mode. - Vortex each cell sample briefly to disperse cell aggregates.
- Place the tube containing a control HeLa-Parkin cell sample that is not infected with mt-Keima lentivirus on the sample injection port and press the "run" button on the cytometer.
- In the dot plot of FSC versus SSC, adjust the FSC and SSC voltage to place cells in the center of the dot plot.
- Draw a P1 gate using the Polygon icon around live cells and eliminate dead cells and cell debris (Figure 2A).
- Adjust voltages for violet and green laser
- Place the tube containing HeLa-Parkin cells infected with mt-Keima lentivirus on the sample injection port and press the "run" button on the cytometer.
- In the dot plot of BV605 (405 nm) versus SSC, adjust the BV605 voltage so that HeLa-Parkin cells expressing mt-Keima are clearly distinguished from control cells that do not express mt-Keima (Figure 2B).
- In the dot plot of PE-CF594 (561 nm) versus SSC, adjust the PE-CF594 voltage so that HeLa-Parkin cells expressing mt-Keima are clearly distinguished from control cells (Figure 2B).
- Assess the percentage of cells in mitophagy
- Acquire the BV605 versus PE-CF594 dot plot using a linear scale. Draw a gate using the Polygon icon around cells expressing mt-Keima, and eliminate cells not expressing mt-Keima (Figure 3A). The mt-Keima fluorescence-positive cell population is referred to as the "mt-Keima" gate.
- Create another dot plot of BV605 versus PE-CF594 showing only "mt-Keima"-gated cells. In this dot plot, draw a gate around untreated mt-Keima-positive cells. The basal mitophagy activity of control HeLa cells is low, and the ratio of emission at PE-CF594/BV605 is less than 1. Thus, this gate is referred to as the "low" gate.
- Draw another gate containing the above region of the "low" gate. This gate is referred to as the "high" gate because it contains cells with high mitophagy activity, that is, cells with a high ratio of emission at PE-CF594/BV605 (Figure 3B).
- To calculate the percentage of cells in the "high" gate, select the "Show Population Hierarchy" menu. The "Percent of Parent" (%Parent) value represents the percentage of cells in the "high" gate among the mt-Keima-positive population.
- Set at least 10,000 events to record in the "mt-Keima" stopping gate and run each sample.
Representative Results
An example of the flow cytometry analysis of CCCP-induced mitophagy in HeLa-Parkin cells is shown in Figure 4. Using the flow cytometry analysis method described above, we can detect a dramatic increase in mitophagic cells in the "high" gate. The percentage of cells in the "high" gate was increased more than 10-fold compared with untreated control cells (Figure 4A). This increase in mitophagy activity was completely abolished by co-treatment with bafilomycin A (BFA) (Figure 4A and 4B).
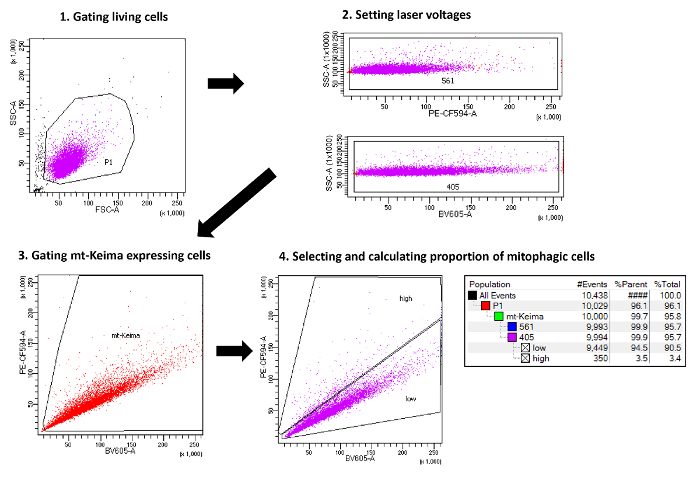
Figure 1. Schematic representation of the method used to measure mitophagy with flow cytometry. The scheme outlines the main steps necessary to quantify cells undergoing mitophagy by flow cytometry. Please click here to view a larger version of this figure.
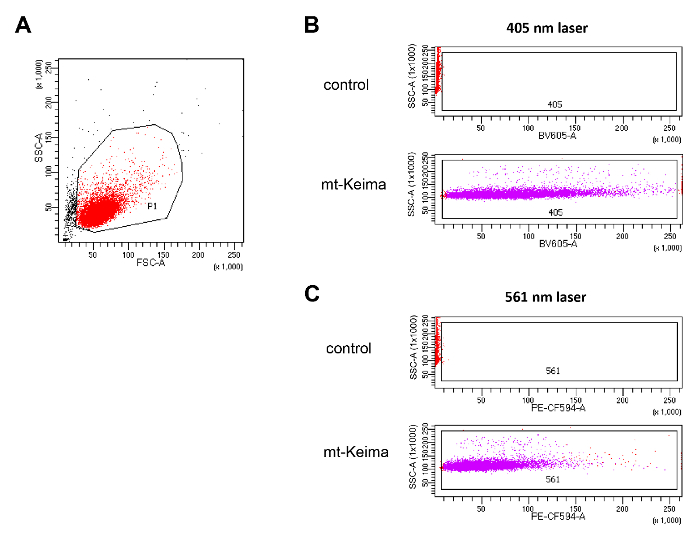
Figure 2. Setting flow cytometer parameters. (A) Gating of living cells. A region for living cells was determined on the FSC versus SSC plot to exclude dead cells and cell debris. (B) Adjusting the 405-nm and 531-nm lasers using control HeLa cells and HeLa cells expressing mt-Keima. Please click here to view a larger version of this figure.
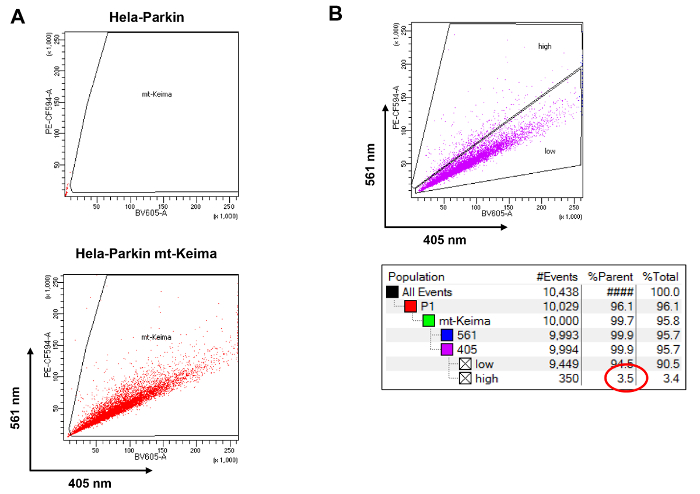
Figure 3. Calculating the proportion of mitophagic cells by flow cytometry. (A) Gating HeLa cells expressing mt-Keima. (B) Gating mitophagic cells and calculating the proportion using the statistics tool. Please click here to view a larger version of this figure.
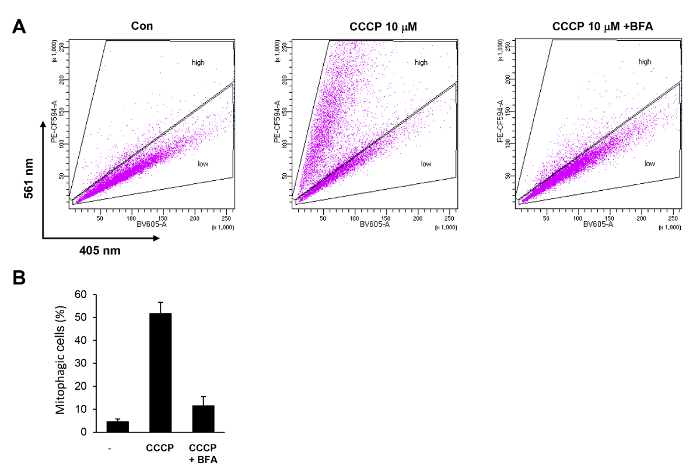
Figure 4. Quantification of CCCP-induced mitophagy in HeLa cells by flow cytometry. (A) HeLa-Parkin cells expressing mt-Keima were treated with 10 µM CCCP for 6 h with or without 100 nM bafilomycin A (BFA). Cells were analyzed by FACS using BV605 and PE-CF594 detectors. (B) Cells with a high PE-CF594/BV605 ratio were selected ("high"), and their proportion was calculated. The results of three repeated experiments are plotted with standard deviations. Please click here to view a larger version of this figure.
Discussion
Here, we present a rapid and sensitive method for using flow cytometry to measure cellular mitophagy activity in cells expressing mt-Keima. Cells undergoing a high level of mitophagy exhibit an increased ratio of PE-CF594 (561 nm)/BV605 (405 nm) excitation. Thus, mitophagy activity can be expressed as the percentage of cells exhibiting a high 561/405 ratio. We calculated the percentage of cells in the "high" gate region on a dot plot of PE-CF594 (561 nm) versus BV605 (405 nm), and the results showed that treatment with CCCP induced an increase in mitophagy in HeLa-Parkin cells. We previously demonstrated that the lentivirus-mediated overexpression of mt-Keima does not itself affect mitochondrial function or cellular physiology31. Although we have shown in our manuscript that flow cytometry analysis can successfully detect CCCP-induced mitophagy in HeLa cells expressing Parkin, this technique can be applied to any type of cell after introducing mt-Keima. We previously showed that hypoxia-induced mitophagy could be detected in HeLa cells without ectopic Parkin expression29. In addition, previous studies, including ours and those of other groups, showed that mt-Keima fluorescence analysis can detect mitophagy in primary cells such as mouse embryonic fibroblasts (MEFs) and immortalized cells such as HeLa, HEK293, and SH-SY5Y cells28,29,32.
Compared with other microscopy-based or immunoblotting-based mitophagy assays, flow cytometry assays have advantages in that they allow rapid, reproducible analyses of large numbers of cells31. Hence, whereas in microscopy-based analyses, researcher bias can distort results, this bias is not an issue when using flow cytometry assays.
Fewer than 1 x 106 cells are required for flow cytometry analysis. In addition, because the analysis of a sample takes only one to two minutes, up to dozens of samples can be analyzed within one to two hours. Furthermore, flow cytometry assays can detect increases in mitophagic cells with good sensitivity. In our flow cytometry analysis, we detected a 10-fold increase in mitophagic cells after 6 h of treatment with CCCP.
The critical step for analyzing mitophagy using flow cytometry is to set the "high" and "low" gates correctly to distinguish cells undergoing a high level of mitophagy. Because basal mitophagy levels can be different depending on the cell type, setting the "high" and "low" gate may be difficult in some cell lines. In such a case, it is helpful to include a control cell line with low basal mitophagy activity such as HeLa cells. In addition, for cells treated with a lysosomal inhibitor such as chloroquine, bafilomycin A can be included as a low mitophagy control to set up the correct gating. Although lentiviruses are commonly used for the ectopic expression of interesting proteins, some cell lines can be susceptible to virus infection. If lentivirus infection itself results in changes in cell growth or mitochondria function, another method should be considered for mt-Keima expression. The expression level of mt-Keima can also vary depending on cell type. If the mt-Keima fluorescence signal is too weak or too heterogeneous, only the cells with a strong mt-Keima fluorescence signal can be collected using a cell sorter or mt-Keima cells treated again with a higher concentration of puromycin antibiotics for several days. The mt-Keima signal of the detached cells are stable as long as the cells are viable, but we recommend analyzing the samples by flow cytometry as quickly as possible.
Our flow cytometry assay is a robust and convenient method for mitophagy analysis, but there are some limitations. First, because the cells should be separated as a single cell level for flow cytometry, this method is difficult to apply for analyzing the mitophagy of tissue or organs without altering the physiology. In addition, mt-Keima fluorescence loses its pH-dependent characteristics after fixation, and thus, all the samples should be analyzed as live cells. This results in the limitation of sample storage mentioned above and another limitation when applying an additional immunofluorescence stain. This technique can measure mitophagy activity quantitatively, but it is unable to monitor changes in mitochondrial morphology or dynamics, which are known to play important roles in the mitophagy process9. Researchers should keep in mind the complexity and undetermined aspects of mitophagy. Therefore, in order to avoid possible misjudgment, the mitophagy process should be further confirmed by the combined results of other common mitophagy analysis methods, including electron microscopy, mitochondrial protein turnover, a reduction in mitochondrial DNA, and fluorescence microscopy measuring the colocalization of mitochondria with lysosomes or autophagosomes as described previously33,34,35. Any observed increase of mitophagy can also be further examined via confocal microscopy of cells expressing mt-Keima. In mitophagic cells, mt-Keima is expressed in a bright punctate pattern with a high 586/440 ratio that can be observed under confocal microscopy28,29.
Given its ability to rapidly and sensitively detect mitophagy, this flow cytometry assay provides a first-step approximation that would be valuable to a variety of studies. By simply infecting cells with the mt-Keima lentivirus, changes of mitophagy activity can be easily measure in any desired cell type. In combination with the cell sorter function, cells with a specific level of mitophagy activity can be specifically isolated and used to study the roles of and regulatory mechanisms underlying mitophagy.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by a grant from National Research Foundation of Korea (2016R1D1A1B03931949) (to J. U.), and by the National Research foundation of Korea (NRF) grant funded by the Korea government(MSIT) (No. 2016R1A2B2008887, No. 2016R1A5A2007009) (to J.Y.)
Materials
| REAGENTS | |||
| poly-L-lysine | Sigma-Aldrich | P2636 | |
| FBS | GIBCO | 16000-044 | |
| penicillin/streptomycin | wellgene | LS202-02 | |
| PBS | Hyclone | SH30013.02 | |
| HEK293T | ATCC | CRL-3216 | |
| DMEM | GIBCO | 12800-082 | |
| OPTI-MEM | GIBCO | 31985-070 | |
| Turbofect | Thermos scientific | R0531 | |
| 0.45 μm syringe filter | sartorius | 16555 | |
| HeLa | ATCC | CCL-2 | |
| polybrene | Sigma-Aldrich | H9268 | 8 mg/ml |
| puromycin | Sigma-Aldrich | P8833 | 2 mg/ml |
| Carbonyl cyanide m-chlorophenyl hydrazine (CCCP) | Sigma-Aldrich | C2759 | 10 mM |
| trypsin-EDTA | wellgene | LS015-01 | |
| EQUIPMENTS | |||
| BD LSRFortessa | BD Bioscience | LSRFortessa | |
| FACSDIVA | BD Bioscience | FACSDIVA (v8.0.1) |
Referências
- McBride, H. M., Neuspiel, M., Wasiak, S. Mitochondria: More than just a powerhouse. Current Biology. 16 (14), R551-R560 (2006).
- Zorov, D. B., Krasnikov, B. F., Kuzminova, A. E., Vysokikh, M., Zorova, L. D. Mitochondria revisited. Alternative functions of mitochondria. Bioscience Reports. Bioscience Reports. 17 (6), 507-520 (1997).
- Taylor, R. W., Turnbull, D. M. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics. 6 (5), 389-402 (2005).
- Galluzzi, L., Kepp, O., Trojel-Hansen, C., Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circulation Research. 111 (9), 1198-1207 (2012).
- Kang, D., Hamasaki, N. Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Current Medicinal Chemistry. 12 (4), 429-441 (2005).
- Sun, N., Youle, R. J., Finkel, T. The mitochondrial basis of aging. Molecular Cell. 61 (5), 654-666 (2016).
- Wallace, D. C., Brown, M. D., Melov, S., Graham, B., Lott, M. Mitochondrial biology, degenerative diseases and aging. BioFactors. 7 (3), 187-190 (1998).
- Yun, J., Finkel, T. Mitohormesis. Cell Metabolism. 19 (5), 757-766 (2014).
- Ashrafi, G., Schwarz, T. L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death and Differentiation. 20 (1), 31-42 (2013).
- Clark, I. E., et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 441 (7097), 1162-1166 (2006).
- Park, J., et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 441 (7097), 1157-1161 (2006).
- Narendra, D., Tanaka, A., Suen, D. F., Youle, R. J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of Cell Biology. 183 (5), 795-803 (2008).
- Zhu, J., Wang, K. Z., Chu, C. T. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy. 9 (11), 1663-1676 (2013).
- Um, J. H., Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Reports. 50 (6), 299-307 (2017).
- Al Rawi, S., et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 334 (6059), 1144-1147 (2011).
- Kim, M. J., et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 12 (8), 1272-1291 (2016).
- Kim, M. J., Yoon, J. H., Ryu, J. H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Reports. 49 (10), 529-535 (2016).
- Sandoval, H., et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 454 (7201), 232-235 (2008).
- Sato, M., Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 334 (6059), 1141-1144 (2011).
- Geisler, S., et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology. 12 (2), 119-131 (2010).
- Matsuda, N., et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. The Journal of Cell Biology. 189 (2), 211-221 (2010).
- Allen, G. F., Toth, R., James, J., Ganley, I. G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Reports. 14 (12), 1127-1135 (2013).
- Chu, C. T., et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature Cell Biology. 15 (10), 1197-1205 (2013).
- Kubli, D. A., et al. PINK1 Is dispensable for mitochondrial recruitment of Parkin and activation of mitophagy in cardiac myocytes. PloS One. 10 (6), e0130707 (2015).
- Dolman, N. J., Chambers, K. M., Mandavilli, B., Batchelor, R. H., Janes, M. S. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy. 9 (11), 1653-1662 (2013).
- Kuma, A., Matsui, M., Mizushima, N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 3 (4), 323-328 (2007).
- Katayama, H., Kogure, T., Mizushima, N., Yoshimori, T., Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chemistry & Biology. 18 (8), 1042-1052 (2011).
- Lee, I. H., Yun, J., Finkel, T. The emerging links between sirtuins and autophagy. Methods in Molecular Biology. 1077, 259-271 (2013).
- Sun, N., et al. Measuring In vivo Mitophagy. Molecular Cell. 60 (4), 685-696 (2015).
- Denison, S. R., et al. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene. 22 (51), 8370-8378 (2003).
- Adan, A., Alizada, G., Kiraz, Y., Baran, Y., Nalbant, A. Flow cytometry: Basic principles and applications. Critical Reviews in Biotechnology. 37 (2), 163-176 (2017).
- Bingol, B., et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 510 (7505), 370-375 (2014).
- Burbulla, L. F., Kruger, R. The use of primary human fibroblasts for monitoring mitochondrial phenotypes in the field of Parkinson’s disease. Journal of Visualized Experiments. 68, e4228 (2012).
- Di Sante, G., Casimiro, M. C., Pestell, T. G., Pestell, R. G. Time-lapse video microscopy for assessment of EYFP-Parkin aggregation as a marker for cellular mitophagy. Journal of Visualized Experiments. 111, e53657 (2016).
- Fang, E. F., et al. In vitro and in vivo detection of mitophagy in human cells, C. Elegans, and mice. Journal of Visualized Experiments. 129, e56301 (2017).
