1. Site-directed Mutagenesis of Protease Vector
- Design and order custom-synthesized oligonucleotides to install a mutation of interest using a modification of standard site-directed mutagenesis protocols12. Standard desalted primers (without additional purification) are perfectly acceptable.
Note: A general approach to primer designs involves creating partially overlapping primers as indicated in Table 1, using a melting temperature (Tm) calculator specific to the polymerase of interest. - Set up polymerase chain reactions (PCRs) for the site-directed mutagenesis on ice, as indicated in Table 2.
- Cycle the PCRs as indicated in Table 3.
- Run 2 – 5 µL of each PCR on a 1% agarose gel and visualize the products to confirm a successful amplification.
Note: A successful PCR will show a single, prominent DNA band at the approximate size marker of the template (~7.8 kB). - Add 0.5 µL (per 25-µL reaction) of DpnI and incubate the samples at 37 °C for 1 h.
- Transform 2 µL of the reaction into chemically competent Escherichia coli.
Note: A fast-growing E. coli strain will reduce incubation times, but any cloning strain will be acceptable. - Plate the transformations onto Luria-Bertani (LB) agar containing 50 – 100 µg/mL carbenicillin. Incubate the plates overnight at 37 °C.
- Select 2 – 4 colonies to grow in a small-scale culture (2 – 5 mL of LB medium with 50 – 100 µg/mL carbenicillin). Incubate the culture at 37 °C at 220 rpm until it is turbid.
- Isolate the plasmid DNA from the cells using a plasmid DNA purification (i.e., “miniprep”) kit according to the manufacturer’s instructions. Quantify the concentration of eluted DNA using a microvolume spectrophotometer.
- Sequence the plasmid DNA extensively using a Sanger sequencing service (such as a core or commercial facility). Prepare the DNA samples according to the service’s specifications. Primers used successfully in prior sequencing reactions are noted in Table 4.
Note: Due to the PCR amplification of the entire plasmid, the sequencing of the entire PCSK9 coding region is recommended for each mutant.
2. High-throughput Luciferase-based Proteolysis Assay
- Cell plating (day 0)
- Use low-passage HEK293T cells for experiments and a culture in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS). Perform all cell work under sterile conditions in a tissue culture hood until ready to assay the luciferase activity. Estimate the number of wells and plates needed for the experiment, anticipating that transfections will be performed in triplicate for each condition tested.
- Dissociate HEK293T cells from a parent flask by treating them with a minimal volume of 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA) to cover the cells. Inactivate the trypsin-EDTA with 2 volumes of DMEM supplemented with 10% FBS and transfer the cells to a sterile tube. Count the cells using an automated cell counter (and staining with trypan blue). Centrifuge the tube at 500 x g for 5 min to recover cells.
- Aspirate trypsin-containing medium and reconstitute the cells in the culture medium to a concentration of 2 x 105 cells/mL. Using a multichannel pipette, transfer 100 µL of the cells to each well of a white (opaque-bottom) 96-well plate, which gives a final seeding concentration of 2 x 104 cells/well.
Note: For initial experiments, it may be useful to additionally seed a sister, clear-bottom 96-well plate, so as to monitor cell growth and adherence during the protocol and guide future troubleshooting. - Incubate the cells at 37 °C and 5% CO2 for 24 h.
- Prepare a master plate of plasmids in a 96-well format. Dilute each plasmid in elution buffer (Tris-HCl, pH 8.5) to 50 ng/µL in an individual well of a 96-well plate.
- Prepare 4 wells each for the positive control (WT) plasmid8, negative control (S386A) plasmid8, and plasmid-free buffer (for mock transfections). If drug or other cellular-based treatments are being planned, then prepare the master plate with the positive control (WT) plasmid in each well, along with 4 wells each of the negative control (S386A) plasmid and the plasmid-free buffer.
- Transfection (day 1)
- Prepare the transfection mixture in a 96-well plate format using deep-well 96-well plates. Perform the transfections in triplicate, being sure to prepare enough reagents to account for pipetting and transfer losses.
Note: The following calculations prepare enough reagent to run one 96-well plate in triplicate (estimated conservatively for 420 wells).- Make a master mix of a diluted lipid transfection reagent, adding 50.4 µL of reagent to 2050 µL of reduced-serum medium. Make a master mix of a DNA precomplexation reagent, adding 33.6 µL of reagent into 1730 µL of reduced-serum medium.
- Using a multichannel pipette, aliquot 16.8 µL of the diluted DNA precomplexation reagent mixture into each well of the deep-well plate.
- Using a multichannel pipette, aliquot 3.2 µL (160 ng) of each plasmid from the master plate into each well of the deep-well plate.
- Using a multichannel pipette, aliquot 20 µL of the diluted lipid transfection reagent mixture to each well of the plate and mix the contents of each well using the multichannel pipette. Cover the plates and let them sit at room temperature (RT) for 10 – 15 min to form lipid:DNA complexes.
Note: Upon transfection, the final components per well will be as follows: 40 ng of DNA, 0.12 µL of transfection reagent, and 0.08 µL of DNA pre-complexation reagent, and the content of each well will be 10 µL in total volume (reduced-serum medium).
- Gently exchange the medium on the 293T cells in the 96-well plates using a multichannel pipette, taking care not to disrupt the cells. Replace the aspirated medium with 95 µL of DMEM supplemented with 10% FBS.
Note: This would be an appropriate time to treat the cells with any drug of interest. - Add 10 µL of the transfection mixture to each appropriate well via a multichannel pipette. Gently swirl the plate to mix the contents in the wells. Incubate the plate at 37 °C with 5% CO2 for 24 h.
Note: The final volume of the wells comes to 105 µL, to account for evaporation over 24 h.
- Prepare the transfection mixture in a 96-well plate format using deep-well 96-well plates. Perform the transfections in triplicate, being sure to prepare enough reagents to account for pipetting and transfer losses.
- Assay (day 2)
- Prepare a stock solution for coelenterazine reagents: 3 M sodium ascorbate [dissolved in phosphate-buffered saline (PBS), prepared fresh], 5 M NaCl, 10 mg/mL bovine serum albumin (BSA; dissolved in PBS, prepared fresh), and 2 mM coelenterazine (dissolved in acidified methanol containing 200 µL of 3 N HCl per 10 mL).
Note: The 2 mM coelenterazine can be stored for 2 weeks when it is kept at -80 °C and in the absence of light. - Prepare 2x coelenterazine reagents for the luciferase readout, with a separate reagent each for the cells and medium, according to Table 5. Mix all reagents save the coelenterazine first, filter the mixture through a 0.22-µm syringe filter, and then add the coelenterazine. Protect the reagents from light until they are ready to be added to the plates.
Note: Due to the loss of solution from filtration, make enough reagent to account for both the loss from filtration, as well as from transfers. Table 5 shows the final concentration of the 2x reagents (as well as the amount of stock solution to add to read out one 96-well plate with one reagent). - Remove the cells from the incubator 24 h after the transfection. Using a multichannel pipette, transfer 50 µL of conditioned medium from each well to a fresh, white-bottomed (opaque) 96-well plate.
- Label the plates as to whether they contain medium or cells. If more than one 96-well plate was transfected, label the plates so as to ensure that each medium-containing plate is paired with its parent plate of cells.
- Using a multichannel pipette, add 50 µL of 2x non-lytic coelenterazine reagent to the plate containing only conditioned medium. Gently rock or shake the plate in the absence of light for 5 – 10 min at RT.
- Using a multichannel pipette, add 50 µL of 2x lytic coelenterazine reagent to the plate containing the cells. Gently rock or shake the plate in the absence of light for 5 – 10 min at RT.
- After the incubation, read out the luminescence of the medium-only plate in a plate reader. Then, read out the luminescence of the cell-containing plate in the same plate reader.
- Discard the plates and save the files for data analysis.
- Prepare a stock solution for coelenterazine reagents: 3 M sodium ascorbate [dissolved in phosphate-buffered saline (PBS), prepared fresh], 5 M NaCl, 10 mg/mL bovine serum albumin (BSA; dissolved in PBS, prepared fresh), and 2 mM coelenterazine (dissolved in acidified methanol containing 200 µL of 3 N HCl per 10 mL).
3. Data Analysis
- Perform an initial data analysis using spreadsheet software. Create a spreadsheet containing the results from the cell and the medium plates.
- Manually inspect the data from the cell plates to identify poorly transfected wells. Wells that show < 5% – 10% of the readout of the negative control (S386A) plasmid should be considered as poorly transfected, making the interpretation of those data suspect.
- Calculate the average background luminescence of each plate from the mock-transfected wells. Subtract the background of each plate from the values of that plate.
Note: This value may be negligible depending on the plate reader used. - Process the data by calculating the proportion of luciferase activity in the medium compared to the overall luciferase activity for each well. Because the cell plate contains both conditioned medium in addition to cells, and the medium-only plate contains the same amount of conditioned medium as the cell plate, it is appropriate to use the following equation:
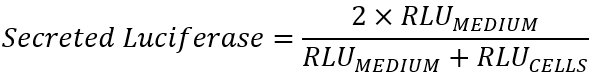
Here, RLU = relative luminescence units, the background-subtracted readout from the plate reader. - Calculate the mean secreted luciferase of the positive control (WT) and the negative control (S386A) wells.
- Evaluate the overall quality of the experiment by calculating a Z-factor13:
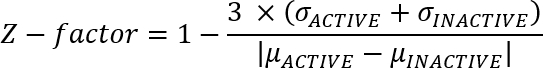
Note: The active values come from the positive control (WT) and the inactive values come from the negative control (S386A) wells. Values closer to 1 indicate a higher experimental quality. Consider repeating the experiment or optimizing the workflow if the value is below 0. - Transfer the data from the spreadsheet program into scientific data analysis software.
- Normalize the secreted luciferase activity to the mean values of the positive control (WT) and the negative control (S386A), setting the positive control as 1 and the negative control as 0.
- Clean the data for outliers using the regression and outlier removal (ROUT) method, setting the maximum false discovery rate to 1%.
- Evaluate for statistically significant differences by comparing the data for each mutant condition (or mutant) to the mean of the WT activity (normalized to a value of 1). Perform multiple unpaired t-tests, correcting for multiple comparisons using the Holm-Sidak method and α = 0.05.
The high-throughput proteolysis assay relies upon overcoming three major challenges. First, to overcome the intrinsically low output of a single-turnover PCSK9 protease, a PCSK9 protease lacking the inhibitory prodomain is used, with the cleavage sequence at the tail of the prodomain linked to a luciferase that can be secreted14. Second, to satisfy the need for the protease to fold in complex with its inhibitory prodomain, the two polypeptides are coexpressed in trans in a cellular context15,16, via a bicistronic vector. Third, to differentiate the cleaved from the uncleaved substrate, the PCSK9 protease is truncated at C678, which prevents the exit of the PCSK9 complex from the endoplasmic reticulum (ER) but has no effect on the proteolytic function17,18. Taken together, this allows for an evaluation of the secreted luciferase as a proxy for the presence and degree of PCSK9 proteolysis (Figure 1A). The general timeframe of the assay is short, with the steps of site-directed mutagenesis of the assay vector (days 1 – 2) followed by the transient transfection (day 3) and luciferase readout (day 4) all completed in as quickly as 4 days, with minimal "hands-on" time (Figure 1B).
Overall, this assay allows for the simultaneous evaluation of PCSK9 proteolysis under a variety of conditions, with the readout done by a luciferase assay. After the acquisition and processing, the data can be visualized in either heat map or tabular format. In particular, mutational analyses of coding SNPs are well suited to this approach. Figure 2 shows a heat map of a saturation mutagenesis library evaluating the cleavage sequence specificity of the PCSK9 protease at substrate sites P6 through P6'. The results show that the optimal cleavage sequence is essentially the same as the WT sequence (SSVFAQ|SIPWNL). In particular, the mutation of the P6 or P4 through P1' residues is poorly tolerated by the protease, consistent with the shallow, hydrophobic binding groove for these positions in the crystal structure of the mature, cleaved PCSK9. From the standpoint of inhibitor development, this narrow sequence specificity profile is a useful finding, as it suggests that no idealized substrate mimetic could outcompete the endogenous cleavage sequence. Figure 3 shows the relative cleavage activity (compared to WT) of a library of missense SNPs described in the clinic, mapped out upon the PCSK9 primary structure. Of the 84 SNPs evaluated, over half showed a significant change in activity compared to the WT protease. These results suggest that alterations in the PCSK9 proteolytic activity are indeed quite common in the clinical population, and such alterations may help to explain the variability in LDL cholesterol levels seen in the clinic.
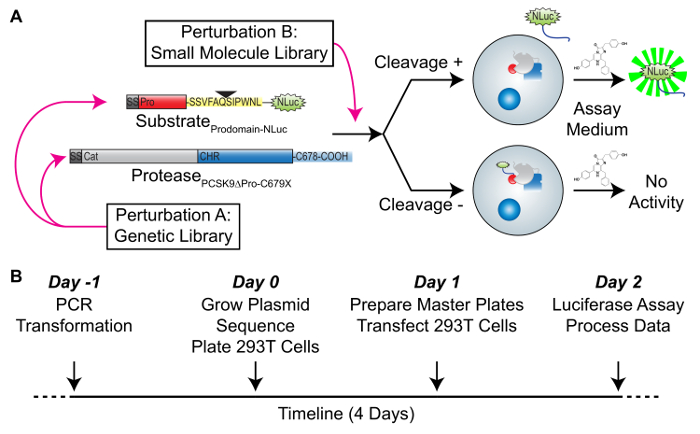
Figure 1: Overall schematic of the high-throughput in trans PCSK9 cleavage assay. (A) This panel shows a biochemical schematic of the assay presented here. The substrate consists of the PCSK9 signal sequence (dark grey) and the prodomain (red) linked to the luciferase that can be secreted (NLuc, green) by the PCSK9 cleavage sequence (yellow). The protease consists of the signal sequence (dark grey), the catalytic domain (light grey), and the cysteine-histidine rich (CHR) domain containing the C679X truncation. The substrate-protease pair is coexpressed at stoichiometric amounts in a 2A-based bicistronic vector, amenable to genetic manipulation. Upon coexpression, the protease and substrate co-fold and remain sequestered in the endoplasmic reticulum (ER). With cleavage activity, the luciferase is freed from the complex and secreted, where it can be detected by the luciferase assay of the conditioned medium. Potential perturbations to the cleavage activity include, but are not limited to, genetic manipulation (perturbation A) or the presence of small molecules (perturbation B). (B) This panel shows the timeline for the overall assay. Site-directed mutagenesis on the WT plasmid is performed on days -1 and 0, followed by the transfection on day 1 and the luciferase assay on day 2, for a total timeline of 4 days. This figure has been adapted from Chorba et al.8. Please click here to view a larger version of this figure.
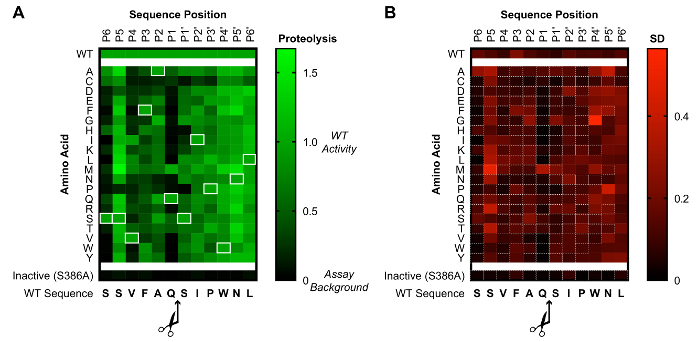
Figure 2: Sequence specificity of the PCSK9 protease. (A) This heat map shows the proteolytic activity, normalized to background and WT, for each single amino acid mutant of a saturation mutagenesis library of the P6 through P6' cleavage sequence. The WT sequence identity is shown at the bottom, with the values for the WT sequence shown both at the top and highlighted by white rectangles. A value of 0 (black) indicates background activity, and a value of +1 (green) indicates the WT value. (B) This heat map shows the standard deviation of the proteolysis values from the same saturation mutagenesis library, with lower values in black and higher values in red. Values outlined in white dashed rectangles represent mutants which showed significantly different proteolysis values from that of the WT protease, as determined by Holm-Sidak-corrected, unpaired t-tests with α < 0.05. The data shown are from 3 independent experiments, each containing 3 replicates. This figure has been adapted from Chorba et al.8. Please click here to view a larger version of this figure.
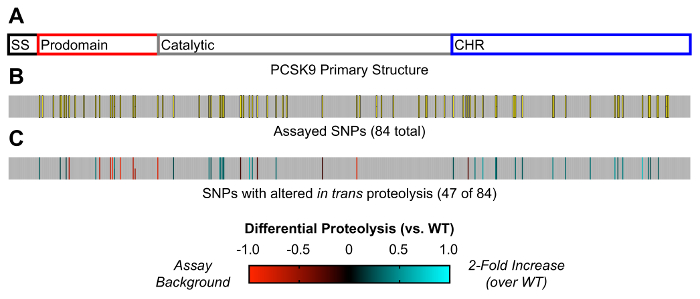
Figure 3: Effects of PCSK9 SNPs on proteolytic function. (A) This panel shows the primary structure of PCSK9, containing the signal sequence (SS; black outline), the prodomain (red outline), the catalytic domain (grey outline), and the cysteine-histidine rich domain (CHR; blue outline). (B) This panel shows the location of 84 clinical SNPs (yellow rectangles with black outlines) placed in the proteolysis assay, mapped onto the primary structure. (C) This panel shows the values of the 47 SNPs with significantly altered proteolytic activity compared to the WT protease, as determined by Holm-Sidak-corrected unpaired t-tests with α < 0.05. The values are mapped onto the PCSK9 primary structure, with a value of -1 (red) indicating no proteolytic activity and a value of +1 (cyan) indicating a 2-fold improvement over the WT protease. This figure has been adapted from Chorba et al.8. Please click here to view a larger version of this figure.
Table 1: Site-directed mutagenic PCR primer design. PCR primers have partially overlapping sequences, according to the design shown. An example of a successful primer pair is shown below the table. Please click here to download this file.
| Component | Stock | Volume (µL) | Final |
| Ultrapure H2O | 17.5 | to 25 µL total volume | |
| Polymerase Reaction Buffer | 5X | 5 | 1X |
| Deoxynucleotides (dNTPs) | 200 µM | 0.5 | 4 µM |
| Template (wild-type (WT)) | 2 ng/µL | 0.5 | 1 ng (total) |
| Primer (Fwd) | 20 µM | 0.625 | 500 nM |
| Primer (Rev) | 20 µM | 0.625 | 500 nM |
| High-Fidelity DNA Polymerase | 2 U/µL | 0.25 | 0.5 U (total) |
Table 2: Components of a PCR for site-directed mutagenesis. The components should be added in the order in which they are listed, with the mixture kept on ice until the reaction begins.
| Step | Temp (°C) | Time | |
| Initial Denaturation | 1 | 98 | 30 s |
| Denaturation | 2 | 98 | 10 s |
| Annealing | 3 | (Tm template)* + 1 | 20 s |
| Extension | 4 | 72 | 30 s/kilobase (kb) (4 min) |
| Cycling | 5 (go to Step 2, 35 cycles total) | ||
| Final Extension | 6 | 72 | 5 min |
| Hold | 7 | 12 | Hold |
| * – Choose the lower Tm template of primer pair | |||
Table 3: Suggested cycling parameters for the PCR. The suggested parameters represent a good starting point but may require optimization.
| Primer | Annealing Location | Sequence (5’ to 3’) |
| 1 | CMV promoter | CGCAAATGGGCGGTAGGCGTG |
| 2 | PCSK9 A95 | TCTCGCAGTCAGAGCGCAC |
| 3 | C-terminus of NLuc | TGTGCGAACGCATTCTGGCG |
| 4 | PCSK9 C301 | GCCAGCGCCTGGCTAGG |
| 5 | PCSK9 E501 | GAGGCCCAAGGGGGCAAG |
Table 4: Validated primers for sequencing. Each primer has been used successfully in sequencing the coding region of the plasmids referenced in this method.
| Component (stock) | Non-Lytic (Medium) | Lytic (Cells) | Amount for 96 wells (1 plate) |
| Sodium ascorbate (3 M) | 300 mM | 300 mM | 700 µL |
| NaCl (5 M) | 5 mM | 5 mM | 7 µL |
| BSA (10 mg/mL, 1%) | 0.10% | – | 700 µL |
| Triton X-100 | – | 0.10% | 7 µL |
| PBS | To 50 µL/well | To 50 µL/well | To 7000 µL |
| Filter through 0.22 µm membrane and transfer 5880 µL of filtrate into fresh tube | |||
| Coelenterazine (2 mM) | 40 µM | 40 µM | 120 µL |
| Total volume: 6000 µL | |||
Table 5: Components of 2x coelenterazine reagents for luciferase readouts. Both non-lytic and lytic reagents are required for each well that is analyzed, so as to separately evaluate the conditioned medium and the transfected cells. The reagents are mixed and filtered prior to the addition of the coelenterazine. After the coelenterazine addition, protect the reagent from light until ready for use.
