Nerve Stem Cell Differentiation by a One-step Cold Atmospheric Plasma Treatment In Vitro
Summary
This protocol aims to provide detailed experimental steps of a cold atmospheric plasma treatment on neural stem cells and immunofluorescence detection for differentiation enhancement.
Abstract
As the development of physical plasma technology, cold atmospheric plasmas (CAPs) have been widely investigated in decontamination, cancer treatment, wound healing, root canal treatment, etc., forming a new research field named plasma medicine. Being a mixture of electrical, chemical, and biological reactive species, CAPs have shown their abilities to enhance nerve stem cells differentiation both in vitro and in vivo and are becoming a promising way for neurological disease treatment in the future. The much more exciting news is that using CAPs may realize one-step, and safely directed, differentiation of neural stem cells (NSCs) for tissue transportation. We demonstrate here the detailed experimental protocol of using a self-made CAP jet device to enhance NSC differentiation in C17.2-NSCs and primary rat neural stem cells, as well as observing the cell fate by inverted and fluorescence microscopy. With the help of immunofluorescence staining technology, we found both the NSCs showed an accelerated differential rate than the untreated group, and ~75% of the NSCs selectively differentiated into neurons, which are mainly mature, cholinergic, and motor neurons.
Introduction
The directed differentiation of NSCs into a certain lineage for tissue transportation is considered one of the most promising therapies for neurodegenerative and neurotraumatic diseases1. For example, catecholaminergic dopaminergic neurons are especially desired in Parkinson's disease (PD) treatment. However, traditional methods to prepare the desired cells for transportation have many drawbacks, such as chemical toxicity, scar formation, or others, which largely hampers the applications of NSCs in regenerative medicine2. Therefore, it is very necessary to find a novel and safe way for NSC differentiation.
Plasma is the fourth state of matters, following solid, liquid, and gas, and it constitutes more than 95% of matters in the whole universe. Plasma is electrically neutral with unbound positive/negative and neutral particles and is usually generated by a high-voltage discharge in the lab. In the last two decades, the application of plasma in biomedicine has attracted huge attention worldwide as the development of cold atmospheric pressure plasma technology. Thanks to this technic, stable low-temperature plasma can be generated in the surrounding air at atmosphere without arc formation and consists of various reactive species, such as reactive nitrogen species (RNS), reactive oxygen species (ROS), ultraviolet (UV) radiation, electrons, ions, and electrical field3. CAPs have unique advantages for micro-organism inactivation, cancer therapy, wound healing, treatment of skin diseases, cell proliferation, and cell differentiation4,5,6,7. In previous work, we demonstrated that cold atmospheric plasma jet can enhance the differentiation of NSCs in both murine neural stem cell C17.2 (C17.2-NSCs) and primary rat neural stem cells, exhibiting a great potential to become a powerful tool for the directed differentiation of NSCs8. Although the mechanism of CAP enhancement of NSC differentiation is not fully understood yet, NO generated by CAPs has been proved to be a key factor in the process. In this work, we aim to provide a detailed experimental protocol of using an atmospheric pressure helium/oxygen plasma jet for the treatment of NSCs in vitro, cell preparation and pretreatment, morphology observation by inverted microscope, and fluorescence microscopy observation of immunofluorescence staining.
Protocol
1. Cell Cultures and Predifferentiation
- Neural stem cell culture and predifferentiation
- Prepare poly-D-lysine-coated coverslips. Put a sterile coverslip (20 mm in diameter) into a 12-well plate. Coat the cover glass with poly-D-lysine, 0.1% w/v, in water (Table of Materials) for better cell adhesion on the coverslips by following the next steps.
NOTE: Optimal conditions must be determined for each cell line and application.- Aseptically coat the surface of the coverslip with poly-D-lysine, 0.1% w/v, in water. Rock gently to ensure an even coating of the coverslip surface.
- After culturing overnight (~12 h) at 37 °C, remove the poly-D-lysine solution and rinse the surface 3x with 1 mL of sterile water.
- Dry the cells at least 30 min before seeding them.
- Prepare poly-D-lysine-coated coverslips. Put a sterile coverslip (20 mm in diameter) into a 12-well plate. Coat the cover glass with poly-D-lysine, 0.1% w/v, in water (Table of Materials) for better cell adhesion on the coverslips by following the next steps.
- Murine neural stem cell C17.2 (C17.2-NSC) culture and predifferentiation
- Incubate C17.2-NSCs in a 25 cm2 flask in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 5% horse serum (HS), and 1% penicillin/streptomycin at 37 °C and 5% CO2 for ~2 – 3 d.
- When the cells reach 85% confluence, remove the medium, wash the cells with 1 mL of PBS and add 1 mL of fresh trypsin (0.25%) into the flask. Leave it for 1 min; then, add 1 mL of culture medium to the flask and pipette up and down several times to ensure a single cell suspension.
- Count the density of the cells in the suspension using a hemocytometer. Calculate the required volume of cell suspension to give a final cell concentration of 2 x 104 cells/mL.
- Seed the C17.2-NSCs on the coated coverslips in the 12-well plate with the density of ~2 x 104 cells per well. Check the cell density under a microscope.
NOTE: For a better result, the optimal cell density must be determined before the immunofluorescence experiment. - Incubate the cells at 37 °C in a cell incubator for 12 h to allow for attachment.
- After attachment, wash the cells 2x with 1 mL of PBS and cultivate the cells with differentiation medium consisting of DMEM/F12 with 1% N2 supplement for 48 h before the plasma treatment.
- Primary rat neural stem cell culture and predifferentiation
- Culture the primary rat NSC suspension in rat NSC growth medium in uncoated T25 flasks at a density of 5 x 105 cells.
NOTE: The primary rat NSCs were isolated following the protocol of Xie et al.9. - Check the formation of neurospheres under an inverted microscope. Make sure the morphology of the neurospheres exhibits spherical and transparent multicellular complexes.
- When the neurospheres reach a diameter of 3 mm or larger, transfer the neurosphere suspension to a 15 mL sterile centrifuge tube and let the neurospheres settle by gravity.
- Remove the supernatant carefully to leave the neurospheres in a minimal volume of medium.
- Rinse the neurospheres 1x with 5 mL of PBS and let the neurospheres settle by gravity. Remove the supernatant to leave a minimal volume of PBS.
- Add a suitable volume of differentiation medium to adjust the cell density to ~12 – 15 neurospheres per milliliter. The differentiation medium for primary rat NSCs consists of DMEM/F12, 2% B27, and 1% FBS.
- Seed 1 mL of neurosphere suspension onto each coated coverslip in the 12-well plate.
NOTE: Shake the plate gently to ensure that the neurospheres are evenly distributed. This step is critical for a better immunofluorescence result. - Place the plate into the incubator and allow it to predifferentiate for 24 h before the plasma treatment.
- Culture the primary rat NSC suspension in rat NSC growth medium in uncoated T25 flasks at a density of 5 x 105 cells.
2. Preparation of the Plasma Jets
- Choose a half-open quartz tube with an internal diameter of 2 mm and an external diameter of 4 mm. Insert a high-voltage wire with a diameter of 2 mm into the tube.
- Insert the quartz tube with the high-voltage wire into a 5 mL syringe and use a holder to mount it inside the center of the syringe. Set the distance between the sealed end of the quartz tube and the syringe tip at 1 cm.
- Connect a 1 m silicone rubber pipe (with an inner diameter of 12 mm) to the open end of the syringe and, then, connect it to the flowmeter and gas valve in sequence.
3. Acquisition of the Jets
- Connect the circuit as shown in Figure 1. Connect the output wire of the power supply to the plasma jet device and, then, connect the tip of the high-voltage probe with the output wire to detect the voltage. Connect the other end of the high-voltage probe to the oscilloscope to record the information of the output voltage. Check the whole circuit and make sure the power supply, the oscilloscope, and the high-voltage probe are all grounded.
- Check the gas line. Make sure the gas tube has been connected to the plasma jet device; then, open the gas valve of the helium (volume fraction, 99.999%) and oxygen (volume fraction, 99.999%) and set the gas flow to 1 L:0.01 L/min (He:O2).
NOTE: Let the gas flow for several minutes before turning on the power supply for the first time. - Set the pulse amplitude, frequency, and pulse width as 8 kV, 8 kHz, and 1600 ns, respectively. Check the circuit again and, then, turn on the output button to create a plasma jet.
CAUTION: Do not touch the high-voltage wire at any time.
4. Plasma Treatment of Neural Stem Cell
- Set the distance between the nozzle of the syringe and the cell well hole to 15 mm.
NOTE: The distance is measured from the bottom of the platform where the 12-well plate is placed. - Take the 12-well plate out of the incubator and change the medium of the predifferentiated cells to 800 µL of fresh differentiation medium.
- Divide the cells into three groups: an untreated control group, a 60 s He-and-O2 (1%) gas flow treatment group, and a 60 s plasma treatment group.
NOTE: There is no obvious liquid loss under the treatment condition. - Place the 12-well plate under the plasma jets, make sure the syringe nozzle is fixed in the center of each hole, and give the relevant treatment to the different groups mentioned in step 4.3.
NOTE: The untreated controls were kept in differentiation medium at room temperature during the experimental procedure to ensure uniform treatment conditions. The He-and-O2 (1%) gas flow treatment group was treated with only a He and O2 (1%) gas flow, without plasma generation. All the treatments should be performed in triplicate.
5. Neural Stem Cell Differentiation
- After treatment, remove the original culture and add 1 mL of new differentiation medium to each well.
- Incubate the cells in the incubator at 37 °C and 5% CO2 for 6 d. Change the medium every other day.
- Check the differentiation status of the different groups daily under an inverted phase-contrast light microscope. Randomly select at least 12 fields and take photos to record the morphological changes.
6. Immunofluorescence Staining
- Rinsing
- Take the samples out of the cell incubator and remove the medium by aspiration. Rinse the cells 1x with 1 mL of PBS.
NOTE: After the differentiation, the cells are easily detached. It is necessary to add the PBS from the side of the culture wells to avoid washing off the cells.
- Take the samples out of the cell incubator and remove the medium by aspiration. Rinse the cells 1x with 1 mL of PBS.
- Fixation
- Fix the cells with 500 µL of 4% paraformaldehyde for 20 min at room temperature. After the fixation, gently rinse the cells 3x with 1 mL of PBS, 5 min each, to remove the residual 4% paraformaldehyde.
NOTE: The optimal time point for cell fixation must be determined according to the cell types. After the fixation, add 1 mL of PBS from the side of the culture wells and leave the sample on the table for 5 min. Do not use a lab shaker, to ensure a minimal loss of cells.
- Fix the cells with 500 µL of 4% paraformaldehyde for 20 min at room temperature. After the fixation, gently rinse the cells 3x with 1 mL of PBS, 5 min each, to remove the residual 4% paraformaldehyde.
- Permeabilization
- Permeabilize the samples with 0.2% TritonX-100 in PBS for 10 min at room temperature. After permeabilization, rinse the cells gently with 1 mL PBS for three times, 5 min each.
NOTE: The optimal time point for cell permeabilization must be determined according to cell types. After permeabilization, add 1 mL PBS from the side of the culture wells, leave the sample on the table for 5 min. Do not use a lab shaker to ensure the minimal loss of cells.
- Permeabilize the samples with 0.2% TritonX-100 in PBS for 10 min at room temperature. After permeabilization, rinse the cells gently with 1 mL PBS for three times, 5 min each.
- Blocking
- Add 1 mL of 10% goat serum in PBS to each sample and leave the sample on the table for 1 h to block any nonspecific interactions.
NOTE: Do not use a lab shaker, to prevent the cells from detaching from the slide. A rinse is not necessary in this step.
- Add 1 mL of 10% goat serum in PBS to each sample and leave the sample on the table for 1 h to block any nonspecific interactions.
- Incubation with primary antibody
- Dilute the primary antibody using primary antibody dilution buffer.
NOTE: For optimal results, the final dilution ratio of the primary antibody must be determined by pretest. The dilution ratios of anti-Nestin (for undifferentiated stem cell), anti-β-Tubulin III (for neuron), anti-O4 (for oligodendrocyte), anti-NF200 (for mature neurons), anti-ChAT (for cholinergic neurons), anti-LHX3 (for motor neurons), anti-GABA (for GABAergic neurons), anti-serotonin (for serotonergic neurons), and anti-TH (for dopaminergic neurons) are 1:80, 1:200, 1:100, 1:100, 1:100, 1:200, 1:100, 1:200, and 1:100, respectively. - Apply 300 µL of diluted primary antibodies to different samples.
- Incubate the cell samples overnight at 4 °C.
NOTE: It is recommended to incubate primary antibodies at 4 °C to reduce the background and nonspecific staining. - Remove the primary antibodies and gently rinse the cells 3x with 1 mL of PBS.
- Dilute the primary antibody using primary antibody dilution buffer.
- Incubation with secondary antibody
- Dilute the Cy3-conjugated or Alexa Fluor 488-conjugated secondary antibodies using 3% goat serum in PBS.
NOTE: For optimal results, the final dilution ratio of the secondary antibody must be determined by pretest. - Apply 300 µL of relevant secondary antibodies to detect each primary antibody.
NOTE: All the subsequent steps need to be performed in the dark to prevent fluorescence quenching. - Incubate the cell sample in the dark for 2 h at room temperature.
- Remove the secondary antibodies and rinse the cells gently with 1 mL of PBS for 10 min at room temperature.
- Dilute the Cy3-conjugated or Alexa Fluor 488-conjugated secondary antibodies using 3% goat serum in PBS.
- Nuclear staining
- Apply 500 µL of Hoechst 33258 working solution to immerse the cell sample.
- Incubate the cell sample in the dark for 8 min at room temperature to label the nuclei.
- Gently rinse the cells 3x with 1 mL of PBS, 10 min each, protected from light.
NOTE: For a minimal loss of cells, the optimal washing condition must be determined empirically.
- Mounting
- Place one drop of mounting medium in the center of the microslide.
- Take out of the coverslips (with samples) using tweezers and carefully position the sample on top of the mounting medium. Avoid air bubbles.
- Remove any excess mounting medium with absorbent paper.
- Fluorescence microscopy observation
- Observe the samples under the fluorescence microscopy equipped with filters for Hoechst 33258, Alexa Fluor 488, and Cy3. For each sample, randomly select at least 8 – 12 fields and record images with a camera.
Representative Results
Cell morphology was observed under the inverted microscope every day after the CAP treatment. Figure 2 shows the ordinary inverted phase-contrast light microscope images of the cell differentiation in both cell lines. The plasma-treated group exhibits an accelerated differentiation rate and a high differentiation ratio compared to the control and gas flow group.
The immunofluorescent results of C17.2-NSCs and primary rat NSCs cultured for 6 d after the treatment are shown in Figure 3 and Figure 4, respectively. Nestin (+, green) decreased, β-Tubulin III (+, red) significantly increased, and O4 (+, green) slightly increased in both cell lines compared to the control group. CAP treatments of 60 s effectively enhanced the C17.2-NSCs into neuronal lineage compared to the control group and the gas flow treatment group. Pure gas flow had no visible effect on the NSC differentiation.
Figure 5 shows the neuronal fate specification in the 60 s plasma treatment group. Strong expressions of NF200 (for mature neuron), ChAT (for cholinergic neuron), and LHX3 (for motor neuron) were observed. GABAergic and serotonergic neurons were rarely seen, while no dopaminergic neurons were detected.
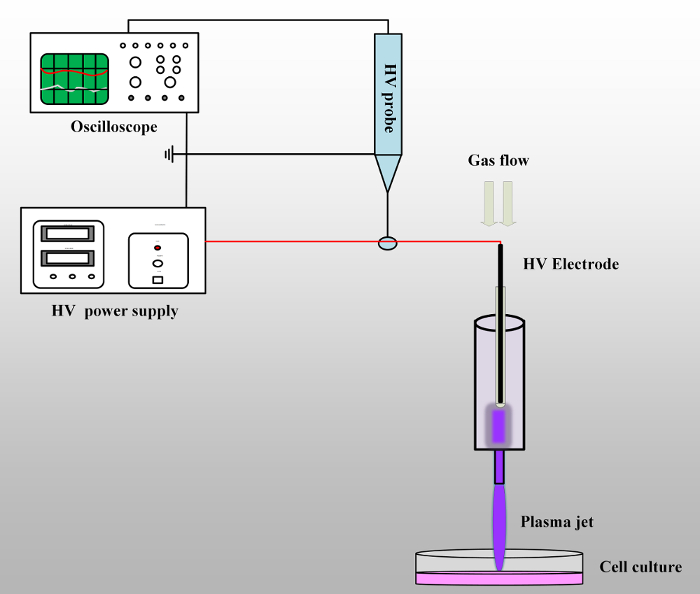
Figure 1: Schematic of the experimental set-up. The plasma jet device is connected to the output wire of the high-voltage power supply. The high-voltage probe with oscilloscope is used to detect the output voltage. When the working gases are flowing through the syringe and the high voltage is on, the plasma jet will be generated and propagate into the open air. Please click here to view a larger version of this figure.
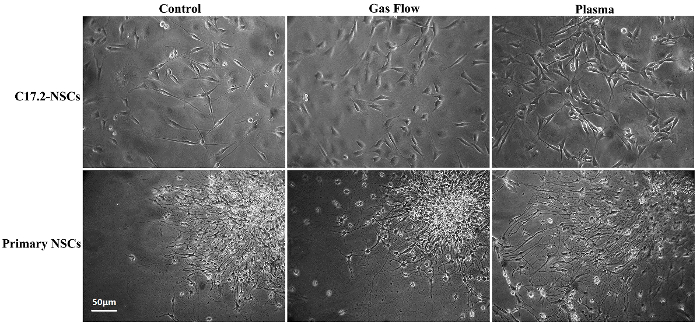
Figure 2: Ordinary inverted phase-contrast light microscope images for C17.2-NSCs and primary rat NSCs. This figure is adapted from Xiong et al.8 with permission. Please click here to view a larger version of this figure.
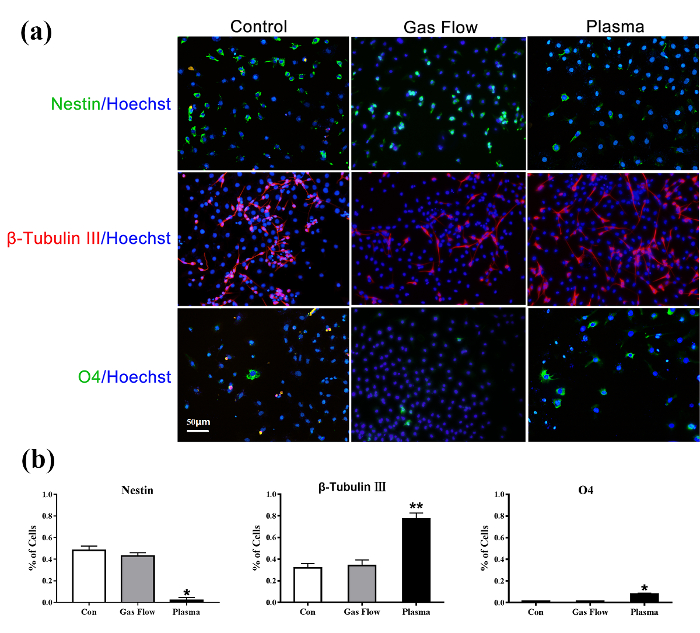
Figure 3: Immunofluorescence detection of untreated C17.2-NSCs (left), 60 s gas flow-treated C17.2-NSCs (middle), and 60 s plasma-treated C17.2-NCSs (right) for 6 d of culture. Nestin (+, green)/Hoechst; β-Tubulin III (+, red)/Hoechst; O4 (+, green)/Hoechst. The nuclear is stained with Hoechst 33258. This figure is adapted from Xiong et al.8 with permission. Please click here to view a larger version of this figure.
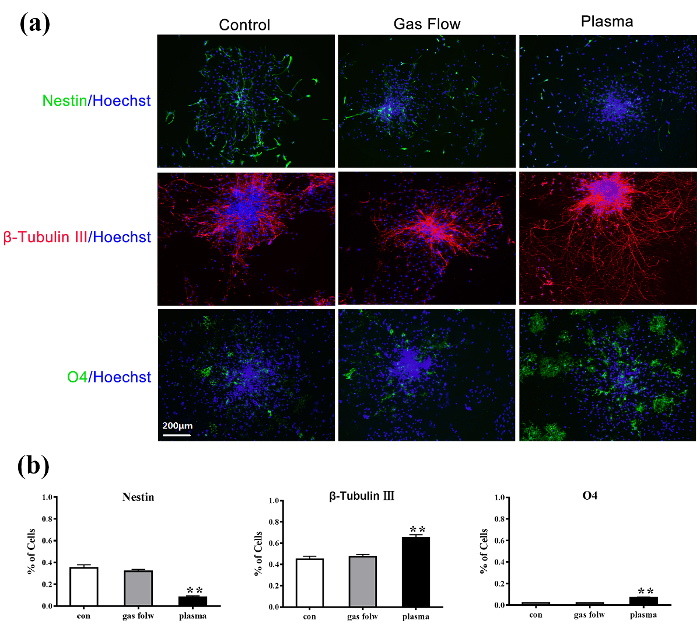
Figure 4: Immunofluorescence detection of untreated primary rat NSCs (left), 60 s gas flow-treated primary rat NSCs (middle), and 60 s plasma-treated primary rat NSCs (right) for 6 d of culture. Nestin (+, green)/Hoechst; β-Tubulin III (+, red)/Hoechst; O4 (+, green)/Hoechst. The nuclear is stained with Hoechst 33258. This figure is adapted from Xiong et al.8 with permission. Please click here to view a larger version of this figure.
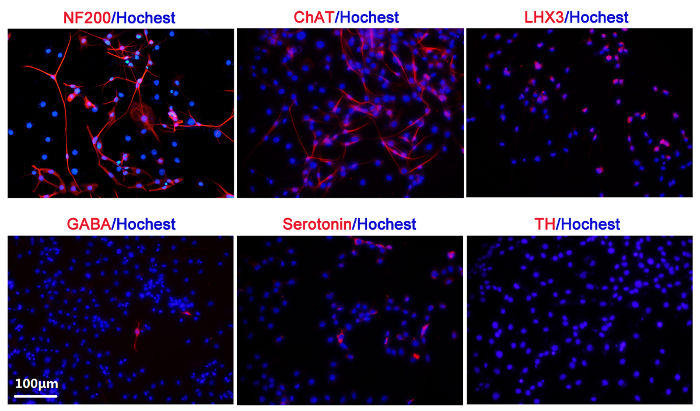
Figure 5: Neuronal fate specification studied by immunofluorescence in the 60 s plasma treatment group. NF200 (+, red)/Hoechst; ChAT (+, red)/Hoechst; LHX3 (+, red)/Hoechst; GABA (+, red)/Hoechst; Serotonin (+, red)/Hoechst; TH (+, red)/Hoechst. The nuclear is stained with Hoechst 33258. This figure is adapted from Xiong et al.8 with permission. Please click here to view a larger version of this figure.
Discussion
C17.2-NSCs is a kind of immortalized neural stem cell line from neonatal mouse cerebellar granular layer cells, developed by Snyder and others10,11. C17.2-NSCs can differentiate into neurons, astrocytes, and oligodendrocytes and are widely used in neuroscience12. In our previous study, CAPs could enhance the differentiation of C17.2-NSCs into neurons. A proof-of-principle study was also carried out using primary rat NSCs, and the effect of the plasma exposure on the primary rat NSCs was qualitatively similar to that of the C17.2-NSCs, with a stronger differentiation of neurons. CAPs, a novel physicochemical technology, may represent a promising tool for neurological disease therapy, such as Alzheimer’s disease, PD, spinal cord injury, and others.
CAP treatment offers a one-step way to enhance both C17.2 NSC and primary rat NSC differentiation in vitro with a short treatment time and little cell damage. Moreover, the results showed a ~75% directed differentiation into neurons, which made plasma treatment a promising method for future tissue transplantations in the clinic. However, the current protocol only recruited one type of CAP devices for NSC differentiation. The limitation of using this microplasma is the nonuniformity when the plasma plume treats the 12-well plate. Future work will consider using a large-volume plasma device or plasma-activated medium for uniform treatment.
There are several critical steps within the protocol. First, each washing step must be carefully performed, for the cells are easily detached after the differentiation. The procedures of fixation and permeabilization are other critical steps in immunostaining. The fixation is necessary to preserve the morphology and antigenicity of the cells13. Permeabilization allows antibodies to bind to the intracellular and nuclear antigens14. The optimal time point for fixation and permeabilization must be determined through pretest. Immunofluorescence blocking and antibody incubation are as important as gentle washing steps. It is recommended to use animal serum from the same source as the secondary antibody to cover the endogenous nonspecific binding proteins. For optimal results, the final dilution ratio of the antibody must be determined by pretest. As for the plasma treatment, the plasma dosage must be applied very carefully; long-time and intense plasma treatment will induce cell apoptosis or necrosis. Therefore, the treatment time and distance must be pretested.
In summary, this manuscript provides a step-by-step protocol for inducing NSC differentiation by using an atmospheric plasma jet and highlights critical issues during the whole process. CAPs can enhance the differentiation of NSCs into neurons and will clearly be beneficial for the treatment of neurological diseases. It is also worthwhile to note that the current protocol only focuses on the in vitro effect of CAPs on NSCs. It is necessary for future studies to evaluate the effect of plasma in vivo using mouse models of nerve injury.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Huazhong Scholar Program, the Independent Innovation Fund of the Huazhong University of Science and Technology (No. 2018KFYYXJJ071), and the National Natural Science Foundation of China (Nos. 31501099 and 51707012).
Materials
| Coverslip | NEST | 801008 | |
| Poly-D-lysine | Beyotime | P0128 | |
| DMEM medium | HyClone | SH30022.01B | stored at 4 °C |
| DMEM/F12 medium | HyClone | SH30023.01B | stored at 4 °C |
| N2 supplement | Gibco | 17502048 | stored at -20 °C and protect from light |
| B27 supplement | Gibco | 17504044 | stored at -20 °C and protect from light |
| Fetal bovine serum | HyClone | SH30084.03 | stored at -20 °C, avoid repeated freezing and thawing |
| Donor Horse serum | HyClone | SH30074.03 | tored at -20 °C, avoid repeated freezing and thawing |
| Penicillin/Streptomycin | HyClone | SV30010 | stored at 4 °C |
| Trypsin | HyClone | 25300054 | stored at 4 °C |
| PBS solution | HyClone | SH30256.01B | stored at 4 °C |
| 4% paraformaldehyde | Beyotime | P0098 | stored at -20 °C |
| TritonX-100 | Sigma | T8787 | |
| Normal Goat Serum Blocking Solution | Vector Laboratories | S-1000-20 | stored at 4 °C |
| anti-Nestin | Beyotime | AF2215 | stored at -20 °C, avoid repeated freezing and thawing |
| anti-β-Tubulin III | Sigma Aldrich | T2200 | stored at -20 °C, avoid repeated freezing and thawing |
| anti-O4 | R&D Systems | MAB1326 | stored at -20 °C, avoid repeated freezing and thawing |
| anti-NF200 | Sigma | stored at -20 °C, avoid repeated freezing and thawing | |
| anti-ChAT | Sigma | stored at -20 °C, avoid repeated freezing and thawing | |
| anti- LHX3 | Sigma | stored at -20 °C, avoid repeated freezing and thawing | |
| anti-GABA | Sigma | stored at -20 °C, avoid repeated freezing and thawing | |
| anti-Serotonin | Abcam, Cambridge, MA | stored at -20 °C, avoid repeated freezing and thawing | |
| anti-TH | Abcam, Cambridge, MA | stored at -20 °C, avoid repeated freezing and thawing | |
| Immunol Staining Primary Antibody Dilution Buffer | Beyotime | P0103 | stored at 4 °C |
| Cy3 Labeled Goat Anti-Rabbit IgG | Beyotime | A0516 | stored at -20 °C and protect from light |
| Alexa Fluor 488- Labeled Goat | Beyotime | A0428 | stored at -20 °C and protect from light |
| Anti-Mouse IgG | |||
| 12-well plate | corning | 3512 | |
| 25 cm2 flask | corning | 430639 | |
| Hoechst 33258 | Beyotime | C1018 | stored at -20 °C and protect from light |
| Mounting medium | Beyotime | P0128 | stored at -20 °C and protect from light |
| Light microscope | Nanjing Jiangnan Novel Optics Company | XD-202 | |
| Fluorescence microscopy | Nikon | 80i | |
| High – voltage Power Amplifier | Directed Energy | PVX-4110 | |
| DC power supply | Spellman | SL1200 | |
| Function Generator | Aligent | 33521A | |
| Oscilloscope | Tektronix | DPO3034 | |
| High voltage probe | Tektronix | P6015A |
Referências
- Temple, S. The development of neural stem cells. Nature. 414 (6859), 112-117 (2001).
- Rossi, F., Cattaneo, E. Neural stem cell therapy for neurological diseases: dreams and reality. Nature Reviews Neuroscience. 3 (5), 401-409 (2002).
- Graves, D. B. The emerging role of reactive oxygen and nitrogen species in redox biology and some implications for plasma applications to medicine and biology. Journal of Physics D: Applied Physics. 45 (26), 263001 (2012).
- Weltmann, K. D., von Woedtke, T. Plasma medicine-current state of research and medical application. Plasma Physics and Controlled Fusion. 59 (1), 014031 (2017).
- Lloyd, G., et al. Gas Plasma: Medical Uses and Developments in Wound Care. Plasma Processes and Polymers. 7 (3-4), 194-211 (2010).
- Yousfi, M., Merbahi, N., Pathak, A., Eichwald, O. Low-temperature plasmas at atmospheric pressure: toward new pharmaceutical treatments in medicine. Fundamental & Clinical Pharmacology. 28 (2), 123-135 (2014).
- Xiong, Z., Roe, J., Grammer, T. C., Graves, D. B. Plasma Treatment of Onychomycosis. Plasma Processes and Polymers. 13 (6), 588-597 (2016).
- Xiong, Z., et al. Selective neuronal differentiation of neural stem cells induced by nanosecond microplasma agitation. Stem Cell Research. 12 (2), 387-399 (2014).
- Xie, Z., Zheng, Q., Guo, X., Yi, C., Wu, Y. Isolation, Culture and Identification of Neural Stem Cells in New-born Rats. Journal of Huazhong University of Science and Technology. [Med Sci]. 23 (2), 75-78 (2003).
- Ryder, E. F., Snyder, E. Y., Cepko, C. L. Establishment and characterization of multipotent neural cell lines using retrovirus vector-mediated oncogene transfer. Journal of Neurobiology. 21 (2), 356-375 (1990).
- Snyder, E. Y., Taylor, R. M., Wolfe, J. H. Neural progenitor cell engraftment corrects lysosomal storage throughout the MRS VII mouse brain. Nature. 374 (6520), 367-370 (1995).
- Snyder, E. Y., Yoon, C., Flax, J. D., Macklis, J. D. Multipotent neural precursors can differentiate toward replacement of neurons undergoing targeted apoptotic degeneration in adult mouse neocortex. Proceedings of the National Academy of Sciences of the United States of America. 94 (21), 11663-11668 (1997).
- Jamur, M. C., Oliver, C. Cell Fixatives for Immunostaining. Methods in Molecular Biology. 588, 55-61 (2010).
- Jamur, M. C., Oliver, C. Permeabilization of Cell Membranes. Methods in Molecular Biology. 588, 63-66 (2010).
