Isolation of Primary Murine Skeletal Muscle Microvascular Endothelial Cells
Summary
Microvascular endothelial cells of skeletal muscles (MMEC) shape the inner wall of muscle capillaries and regulate both, exchange of fluids/molecules and migration of (immune) cells between muscle tissue and blood. Isolation of primary murine MMEC, as described here, enables comprehensive in vitro investigations of the "myovascular unit".
Abstract
The endothelial cells of skeletal muscle capillaries (muscle microvascular endothelial cells, MMEC) build up the barrier between blood stream and skeletal muscles regulating the exchange of fluids and nutrients as well as the immune response against infectious agents by controlling immune cell migration. For these functions, MMEC form a functional "myovascular unit" (MVU), with further cell types, such as fibroblasts, pericytes and skeletal muscle cells. Consequently, a dysfunction of MMEC and therefore the MVU contributes to a vast variety of myopathies. However, regulatory mechanisms of MMEC in health and disease remain insufficiently understood and their elucidation precedes more specific treatments for myopathies. The isolation and in-depth investigation of primary MMEC functions in the context of the MVU might facilitate a better understanding of these processes.
This article provides a protocol to isolate primary murine MMEC of the skeletal muscle by mechanical and enzymatic dissociation including purification and culture maintenance steps.
Introduction
Via bloodstream, cells and organs are supplied with oxygen, substrates and other necessary molecules. This interchange takes place in capillaries, the smallest vessels. Capillaries are formed by an inner endothelial cell (EC) layer whose integrity remains a prerequisite to successful regulation of muscle homeostasis between the intravascular and interstitial space. To ensure a selective transition of soluble factors and cells, EC constitute a monolayer interconnected by tight and adherens junctions 1. Besides its role as barrier for nutrients or metabolic products, EC regulate the recruitment of leukocytes in inflammatory processes. Inflammation or tissue damage leads to an up-regulation of adhesion molecules on the EC surface and production of chemokines facilitating leukocyte attachment and transmigration into the target tissue 2. Consequently, EC are critically involved in the regulation of inflammatory processes such as the defense against pathogens or tissue repair.
A dysfunction of EC is directly associated with vascular diseases, chronic kidney failure, venous thrombosis severe pathogen infections. Furthermore, EC are virtually always involved in organ-specific autoimmunity such as diabetes mellitus or multiple sclerosis 3. The barrier function between blood stream and organs is therefore controlled by a concerted interplay of different cell types. In the skeletal muscle microvascular endothelial cells (MMEC) together with muscle cells, fibroblasts and pericytes form a functional unit, the "myovascular unit" (MVU). Therefore, a dysfunction of the MVU might play a critical role in the pathophysiology of myopathies. However, a deeper understanding of these regulatory mechanisms is still missing and currently precludes the identification of new, urgently needed, therapeutic targets in myopathies.
To investigate the complex physiological and pathophysiological mechanisms, animal models are commonly used. However, in vitro models offer the advantage to focus on the subject of interest by excluding a variety of confounding factors. To investigate processes in vitro it is necessary to isolate pure and viable primary cells. In contrast to cell lines, primary cells isolated from transgenic animals enable to investigate the consequences of genetic modifications in vitro.
Here, a method to isolate primary murine MMEC is described by using mechanical and enzymatic dissociation followed by magnetic activated cell sorting techniques (MCS) for purification. For this purpose, magnetic beads against specific surface markers are used. Platelet endothelial cell adhesion molecule-1 (PECAM1, CD31) is mainly expressed on EC and can be used to enrich this cell type. To warrant high cell purity, cells of hematopoietic origin are excluded by a negative selection for protein tyrosine phosphatase receptor type C (PTPRC, CD45). Further, quality controls, cultivation of primary murine MMEC, potential applications and limitations as well as special considerations are presented.
Protocol
All animal experiments were approved by the local authorities and conducted according to the German animal welfare act (84-02.05.20.13.097).
1. General Remarks on Animal Experiments
- Perform all mouse experiments in accordance with the guidelines of the respective institutional animal care and use committee.
- Keep mice under standardized conditions and according to international guidelines such as the Federation for Laboratory Animal Science Associations (FELASA).
NOTE: In general this isolation technique can be used for mice independent of age, gender or genetic background. To obtain a sufficient cell number, 4–10 week old males are preferred, because biological properties can vary with age and gender.
2. Preparation of Solutions, Media and Coating
- Prepare digestion solution (DS) by mixing 2.2 mL of Dulbecco´s Modified Eagle Medium (DMEM) with 200 μL Collagenase-Dispase and 45 μL Desoxyribonuclease (DNase).
- Prepare 500 mL of Endothelial cell medium (ECM) by mixing 450 mL of DMEM with 50 mL FCS (approximately 10%), 0.25 mL basic fibroblast growth factor bFGF (20 μg/mL; approximately 0.05%) and 5 mL penicillin-streptomycin (approximately 1%). Perform sterile filtration in a glass tube (diameter of filter pores: 0.2 μm). Afterwards store the solution at 4 °C.
- Coat cell culture plates as described below.
- For cultivation of freshly isolated primary murine MEC cover the whole surface with 1 mL per well of a 6 well cell culture plate with a gelatin based speed coating solution (see Table of Materials) according to manufacturer’s instructions for 3–5 min at room temperature (RT).
- Aspirate and discard coating solution. Rinse each well with 2 mL sterile phosphate buffered saline (PBS), pH 7.1–7.5 in two consecutive washing steps.
- Aspirate and discard PBS. Fill up wells with 1 mL volume of ECM.
3. Isolation of Primary Murine Muscle Microvascular Endothelial Cells (MMEC)
- Euthanize one adult 4–12 weeks old, male mouse by cervical dislocation without any anaesthesia. The aim is to quickly separate the spinal cord from the brain so as to provide the animal with a fast and painless death.
- Sever the extremities.
- Use a surgical sharp/blunt (cutting edge 42 mm, straight) and sharp/sharp (cutting edge 23 mm, straight) scissor, a straight (serrated, 2 mm x 1.25 mm x 12 cm) and a curved (serrated, 1.3 mm x 1 mm x 13 cm) forceps.
- Disinfect all surgical instruments with 70% ethanol.
- Position the animal on its back, moisten the legs with 70% ethanol. Sever the whole leg by cutting at the hip joint with the sharp/blunt surgical scissor. Place the extremities in a closed cell culture dish (35 mm x 10 mm).
NOTE: From now on perform every step under a sterile laminar flow hood and work with sterilized instruments.
- Isolate muscle tissue (preferentially use the musculus quadriceps femoris or musculus triceps surae from both legs).
- Cut the skin open from the hip to the toe-tip by using a sharp/sharp scissor and curved forceps. Hold the toe or footpad with the straight forceps. Peel off the skin with the curved forceps from the toe to the hip.
- Isolate the musculus quadriceps femoris by cutting the tendon from the knee and sever the muscle along the femur to the hip. Isolate the musculus triceps surae by severing the Achilles tendon. Hereafter, cut along the tibia to the popliteal fossa and remove the muscle.
NOTE: If large vessels (A. femoralis, A. tibialis anterior, A. tibialis posterior, A. fibularis) are visible in the isolated muscles, remove vessels to avoid contamination by macrovascular endothelium. - To remove large vessels, hold the part of muscle not containing large vessels with a curved forceps and cut it off next to the vessel. For this protocol, 1 g or less muscle tissue equal to both quadriceps femoris and triceps surae is recommended.
- Add 2,445 µL DS (from step 2.1) to a cell culture dish (35 mm x 10 mm) and determine the weight.
- Transfer all muscle pieces to this cell culture dish containing the 2445 µL DS and determine the weight. The difference of both measured values provides the dry weight of muscle tissue which must not exceed 1 g.
- Cut the whole muscle tissue into small pieces (≤2 mm cubes, about 100 pieces) by using the sharp/sharp scissor.
- Dissociate the muscle tissue.
NOTE: This step takes about 120 min.- Store muscle/DS suspension at 37 °C (incubator with 5% CO2) for 1.5 h. Mix the suspension carefully every 20 min for about 5 min using a 1 mL insulin syringe.
- Transfer suspension to a 70 μm nylon cell strainer placed on a 50 mL tube and collect the flow through. Wash the cell strainer with 8 mL of DMEM and collect the flow through.
- Discard cell strainer and centrifuge suspension for 10 min at 300 x g at 20 °C. Carefully remove supernatant.
CAUTION: pellet is easy to lose. - Resuspend cell pellet in 1 mL of an Ammonium-Chloride-Potassium (ACK) lysing buffer (see enclosed Table of Materials) for the lysis of red blood cells and incubate for 30 s at RT. Add 9 mL DMEM + 10% FCS to stop the reaction and transfer cell suspension to a 15 mL tube.
- Deplete CD45+ cells following the CD45 microbeads manufacturer’s instructions. For all following steps of CD45+ depletion use MCS buffer (see enclosed Table of Materials).
NOTE: This step takes about 60 min.- Determine cell numbers using a Neubauer cell counting chamber (expect about 5 x 106 cells per g muscle tissue).
- Centrifuge suspension at 300 x g for 10 min at 4 °C. Take off supernatant completely and resuspend cell pellet in 90 µL MCS buffer (per 107 cells or less). Add 10 μL of CD45 microbeads (per 107 cells or less).
- Mix cell suspension and incubate for 15 min in the fridge at 4–8 °C.
- Add 1mL MCS buffer (per 107 cells or less). Centrifuge at 300 x g for 10 min at 4°C. Remove the supernatant completely and resuspend cells in 500 μL MCS buffer.
- For magnetic cell separation use a large magnetic column (LMC) with a reservoir volume of 8 mL and a capacity of up to 2 x 109 total cells. Position the LMC in the magnetic field of the separator.
- Rinse with 3 mL of MCS buffer into the reservoir of the LMC. After rinsing, place a 15 mL conical tube below the column to collect flow through. Apply the whole cell suspension (500 µL) onto the column and let it completely flow through.
- Wash the column three times by adding 3 mL of MCS buffer into the reservoir and wait until the column´s reservoir is empty before performing the next washing step.
- Collect the unlabeled cells passing the column use for further separation steps (representing the CD45– fraction). Labeled cells (representing the CD45+ fraction) accumulated in the column can be discarded.
- Accumulate CD31+ cells following CD31 microbeads manufacturer’s instructions. For all following steps of CD31+ accumulation use MCS buffer.
NOTE: This step takes about 60 min.- Determine cell number using a Neubauer cell counting chamber (expect about 4 x 106 cells per g muscle tissue).
- Centrifuge obtained unlabeled cell suspension from step 3.5 for 10 min at 300 x g at 4°C.
- Remove the supernatant completely. Resuspend the pellet in 90 μL MCS buffer and add 10 μL of CD31 microbeads (per 107 total cells or less). Mix the whole suspension and incubate for 15 min in the fridge at 4–8 °C.
- Add 1mL MCS buffer and centrifuge at 300 x g for 10 min at 4 °C. Take off the supernatant and resuspend cells in 500 µL of MCS buffer.
- For the magnetic cell separation use a medium magnetic column (MMC) with a reservoir volume of 3.5 mL and a capacity of up to 2 x 108 total cells.
- Position the MMC in the magnetic field of the separator. Rinse the MMC with 500 µL of MCS buffer. After rinsing, place a 15 mL conical tube below the column to collect flow through.
- Apply the whole cell suspension (500 µL) onto the column and let it completely flow through.
- Wash the column three times by adding 500 µL of MCS buffer and wait until the column’s reservoir is empty before performing the next washing step. Unlabeled cells passing the column represent the CD45– CD31– fraction. Store them for further quality controls.
- Remove column from the separator and place it on a suitable collection tube (15 mL tube). Pipette 2 mL MCS buffer onto the column. Immediately flush out the magnetically labeled cells by firmly pushing the plunger into the column. This CD45– CD31+ fraction represents the enriched primary murine EC.
- Centrifuge cell suspension for 5 min at 350 x g at 20 °C. Remove supernatant completely and resuspend cells (expect around 0.6 x 106 cells per g muscle tissue) in 1 mL ECM (see step 2.2.). Transfer cells (0.5–0.7 x 106 cells) to a single well of a coated 6-well culture plate (from step 2.3) containing 1 mL of ECM.
- For cultivation, incubate cells at 37 °C and 5% CO2 in a sterile incubator. Refresh the ECM every two to three days.
4. Primary Murine MMEC Purification
- Perform a second cycle of CD31+ accumulation, following manufacturer’s instructions (CD31 microbeads mouse protocol; see step 3.6.).
- Detach cells with Trypsin/EDTA solution when they are at 80–90% confluence (usually after 7 days). For this, rinse each well with 2 mL sterile PBS, in two consecutive washing steps. Use 800 μL Trypsin/EDTA solution per 6-well and incubate at 37 °C and 5% CO2 in a sterile incubator for 3–5 min. Stop enzymatic activity by using 1200 μL DMEM containing a minimum of 10% FCS.
- Centrifuge the cell suspension for 5 min at 350 x g at 20 °C.
- Perform the second CD31-MCS step to increase the purity (according to the CD31 microbeads manufacturer´s instructions; see 3.6.).
- Observe cell confluence via bright field or standard phase-contrast microscopy using a
20x magnification and 0.35 lens numerical aperture. See Figure 1.
NOTE: Cells can be used for respective experiments or kept in culture by passaging. Use cells for experiments promptly in lower passages. It is not recommended to use cells after passage
8–10.
5. Quality Control
- Perform flow cytometry of primary murine MMEC after the second CD31-MCS-step.
- Prepare a FACS tube by adding 1 mL flow cytometry (FC) buffer (see enclosed Table of Materials).
- Detach cells with Trypsin/EDTA solution when they are at 100% confluence (usually after 14 days). For this, rinse each well with 2 mL sterile PBS, in two consecutive washing steps. Use 800 μL Trypsin/EDTA solution per 6-well and incubate at 37 °C and 5% CO2 in a sterile incubator for 3–5 min. Stop enzymatic activity by using 1200 μL DMEM containing 10% FCS.
- Determine cell number using a Neubauer cell counting chamber and add a minimum of 1 x 105 cells to the prepared FACS tube.
- Centrifuge cell suspension for 10 min at 300 x g at 4 °C. Perform two washing steps by removing the supernatant, resuspending cells in 1 mL FC buffer and centrifuging for 10 min at 300 x g at 4 °C.
- Prepare a staining mix by adding anti-mouse CD31-FITC antibody (1:100) and anti-mouse CD45-PE (1:100) with FC buffer.
- Resuspend cells in 100 μL of staining mix in the FACS tubes. Incubate for 30 min at 4 °C. Add 1 mL of FC buffer. Centrifuge cell suspension for 5 min at 300 x g at 4 °C. Carefully remove supernatant and resuspend cells in 200 μL of FC buffer.
- Measure cells in a flow cytometer following the gating strategy is shown in Figure. 2A.
- Alternatively perform immunofluorescence staining of primary murine MMEC after the second CD31-MCS-step.
- Coat the coverslips.
- Transfer a sterile 13 mm diameter round glass coverslip into a well of a 24 well plate. Coat coverslip by adding 300 µL of speed coating solution per well. Incubate for 3–5 min at room temperature (RT).
- Aspirate and discard coating solution. Rinse each well with 2 mL sterile PBS in two consecutive washing steps. Aspirate and discard PBS.
- Seed 1 x 105 cells in 1 mL ECM on the coated coverslip and incubate at 37 °C and 5% CO2 in a sterile incubator until cells are 99% confluent.
- Perform fixation and immunofluorescence staining.
- Remove the ECM medium of cultivated cells from 5.2.1.3.
- Fix cultured cells by adding 300 µL 4% paraformaldehyde (PFA) with pH of 7.4 per well, for 10 min at 4°C.
CAUTION: PFA is toxic and must be handled with care. - Perform 3 washing steps by adding 1 mL PBS per well and remove supernatant after 5 min at RT.
- Prepare a blocking solution containing 5% BSA, 0.2% Triton-X and 1% goat serum in PBS.
- Add 300 µL of blocking solution to each well and incubate for 1 h at RT.
- Remove the supernatant and add 200 µL per well of an anti-PECAM-1 (CD31) antibody (1:200) in 5% BSA, 1% goat serum in PBS and incubate at 4 °C overnight.
- Perform 3 washing steps by adding 1 mL PBS per well and removing supernatant after 5 min at RT in each step.
- Prepare a secondary antibody solution containing Cy3-conjugated anti-rat IgG antibody (1:500) in 1% BSA and PBS. Add 300 µL per well of the secondary antibody solution and incubate for 1 h at RT in the dark.
- Perform 3 washing steps by adding 1 mL PBS per well and removing supernatant after 5 min at RT in each step.
- Take out the round glass coverslips carefully with forceps and transfer it onto a microscopy slide. Add an appropriate amount of mounting medium containing DAPI on the cell layer and carefully put a cover glass on it (see enclosed Table of Materials).
- Observe cells with an appropriate fluorescence microscope equipped with a fluorescence light source (120 W) and two filter sets: An excitation/emission filter set with 550/25 nm and 605/70 nm wavelengths to detect Cy3 540/562 nm.
- Use a second filter set with an excitation/emission wavelength of 365/50 nm and 445/50 nm to detect DAPI 350/440 nm. Use a magnification of 40x and 80% intensity of 14.5 mW/cm2 for DAPI with an acquisition duration of 30 ms. For detection of Cy3 use 80% of 67.5 mW/cm2 with an acquisition duration of 60 ms.
- Coat the coverslips.
- Perform quantitative PCR.
- Isolation of mRNA.
- Follow standard procedures using an acid guanidinium thiocyanate-phenol (AGTP) reagent to isolate mRNA. Use a minimum of 0.5 x 106 cells from CD45– CD31– (see step 3.6.8), CD45– CD31+ (see step 3.6.9.) fractions and cultivated primary murine MMEC (4.1.3)
- Centrifuge cell suspension for 10 min at 300 x g at 20 °C. Take off the supernatant completely. Resuspend the cell pellet in 500 µL of AGTP reagent and transfer cell suspension into a 2 mL reaction tube. Incubate for 5 min at RT.
- Add 100 µL of chloroform and shake vigorously for 15 s. Incubate for 3 min at RT.
- Centrifuge suspension for 15 min at 12,000 x g at 4°C.
- Take off the upper aqueous phase (containing RNA) carefully and transfer into a 1.5 mL reaction tube. Add 250 µL of isopropanol and incubate for 10 min at RT.
- Perform a centrifugation step at 12,000 x g for 10 min at 4 °C.
- Take off the supernatant and add 1 mL of 75% ethanol carefully. Centrifuge at 7,500 x g for 5 min at 4 °C.
NOTE: Pellet is easy to lose. - Remove the supernatant completely. Let the pellet dry on air for 5–10 min.
NOTE: ethanol needs to be removed but do not dry too much. - Dissolve pellet in 30 µL diethylpyrocarbonate treated water containing in and heat for 10 min at 55 °C.
NOTE: RNA concentration and purity can be measured by a spectral-photometer.
- Perform cDNA synthesis (reverse transcriptase PCR).
- Prepare a mastermix containing: 10 µL of PCR buffer (5x), 2,5 µL desoxyribonucleosid-triphosphate (dNTP (10 mM)), 0.5 µL random mixture of single-stranded primer (0.2 µg/ µL), 1 µL of RNase inhibitor (40 U/ µL, 0.4 reverse transcriptase (200 U/µL) and 5.6 µL of diethylpyrocarbonat treated water (see enclosed Table of Materials).
- Dilute 500 ng RNA in 30 µL diethylpyrocarbonate treated water in 0.2 mL PCR tubes and add the whole 20 µL mastermix.
- Use a PCR cycler performing following steps: 10 min 25 °C, 30 min 50 °C, 5 min 85 °C and hold at 4 °C.
- Isolation of mRNA.
- Perform Quantitative PCR (qPCR).
NOTE: Perform qPCR of samples in duplicates or triplicates. - Use primer with two different fluorescence dyes. Primer with an absorption of 495 nm and an emission of 517 nm for the gene of interest.
- For detection of the satellite cell marker gene expression, use primers for paired box protein 7 (Pax7) and M-cadherin (Cdh15) (see enclosed Table of Materials).
- For detection of the endothelial cell marker gene expression, use primers for claudin-5 (Cld5), occludin (Ocln) and zonula occludens-1 (Tjp1 or ZO1) (see enclosed Table of Materials).
- As reference gene for 18s rRNA, use a primer with absorption of 538 nm and an emission of 554 nm wavelength.
- Prepare a qPCR mastermix for each gene of interest with the respective primer. Mastermix contains: 10µl qPCR buffer (2x), 1 µL of Primer for gene of interest (10 µM), 1 µL of Primer for 18s rRNA and 4 µL of nuclease free water.
- Add 16 µL of the mastermix to a single well of a 96-well reaction plate. Add 4 µL of cDNA (obtained from step 5.3.2.) per well containing the mastermix.
- Use a real-time PCR cycler performing following steps: holding stage with 50 °C for 2 min and 95 °C for 10 min. Cycling stage with 40 cycles of 95 °C 15 s and 60 °C for 1 min.
Representative Results
One day after isolation, primary murine MMEC and residual other cells form conglomerates and adhere to the bottom of culture dishes (Figure 1A day 1). From day 7 on, flat and elongated cells can be observed. However, contamination of other, mostly spheroid cells, is still visible (Figure 1A day 7). Thus, another cycle of CD31 positive selection via MCS is required. Hereafter, primary murine MMEC proliferate to a density of approximately 80–90%. Upon confluence they typically form a non-overlapping monolayer of longitudinally aligned cells (Figure 1A day 14). Proliferation stops upon confluence due to contact-inhibition. After 14 days about 5–10 x 105 cells can be used for further investigations.
Quality control via flow cytometry using fixable viability dye FVD780 (to stain for dead cells), PECAM1 (CD31) and PTPRC (CD45, as marker for cells of hematopoietic origin) showed values for both viability and purity ranging around 70% each for cells immediately after the isolation (Figure 2A). Cells cultivated after another CD31 positive selection via MCS, showed satisfying values for purity as well as for viability ranging up to 95% each (Figure 2B).
To evaluate the accuracy of the selection steps, obtained cells were further investigated for gene expression of the muscle satellite cell marker genes paired box protein 7 (Pax7) and M-cadherin (Cdh15) on mRNA level by quantitative PCR (qPCR). Primary murine MMEC (pmMMEC) and differentiated primary murine muscle cells (pmMC) were used as negative and positive control, respectively. Murine muscle cells were commercially purchased. As expected, only the CD45– CD31– fraction as well as the pmMC expressed Pax7 and Cdh15, whereas CD45– CD31+ and the primary murine MMEC were negative for these markers (Figure 2C).
EC derived from capillaries in the endomysium of skeletal muscle express tight junction proteins 4,5. By using qPCR, the expression of claudin-5 (Cld5), occludin (Ocln) and zonula occludens-1 (Tjp1 or ZO1) of confluent primary murine MMEC after the second CD31 MCS step was evaluated. PmMC were used as controls (Figure 2D). Primary murine MMEC expressed high levels of Cld5, Ocln and Tjp1, whereas pmMC only show low expression of Tjp1. Furthermore, immunofluorescence staining as quality control confirmed surface expression of the endothelium-specific marker PECAM1 in primary murine MMEC (Figure 2E).
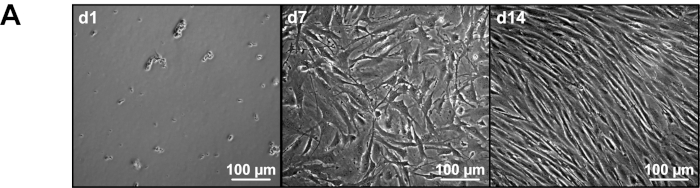
Figure 1: Morphology of primary murine MMEC. (A) Representative image of cultured primary murine MMEC by phase contrast microscopy from days 1 to 14. d1 = Day 1, d7= Day 7, d14 = Day 14. Please click here to view a larger version of this figure.
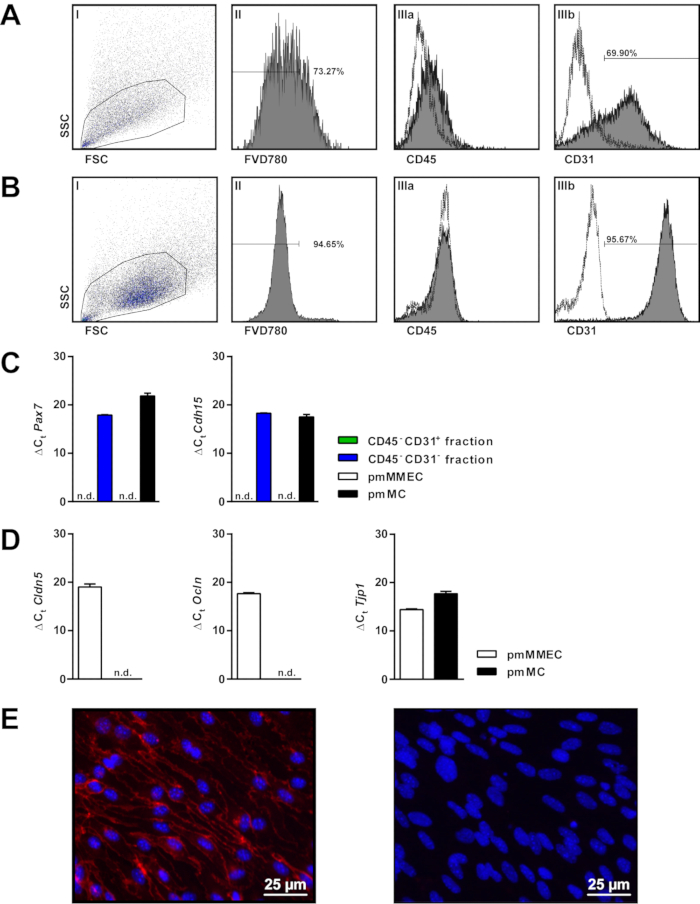
Figure 2: Quality control of primary murine MMEC. Gating strategy for flow cytometric analysis of primary murine MMEC immediately after isolation (A) and on day 15 (B) using (I) FSC/SSC, (II) FVD780 and (IIIa) CD45, (IIIb) CD31 (dashed lines = isotype control). (C) Expression level of Pax7 and M-Cadherin (Cdh15) in CD45– CD31–, CD45– CD31+ fractions after MCS isolation as well as in primary murine MMEC (pmMMEC) and primary murine muscle cells (pmMC). Expression levels are shown as ΔCt values (sample – 18S rRNA), n.d. = not determined. (D) Expression level of tight junction proteins claudin-5 (Cldn5), occludin (Ocln) or zonula occludens-1 (Tjp1 or ZO-1) of pmMMEC and pmMC, shown as ΔCt values. (E) Immunofluorescence staining for PECAM1 (red) in cultivated primary murine MMEC 24 h after second CD31 positive selection. Nuclear staining with DAPI-containing (blue) mounting medium; left = anti-PECAM1/DAPI, right: negative control/DAPI. Please click here to view a larger version of this figure.
Discussion
Microvascular endothelial cells provide barrier functions in all tissues and their dysfunction results in disease of the associated organs 3. Moreover, organ-specific studies of microvascular EC could pave the way for new therapeutic strategies. Therefore, a deeper understanding of microvascular EC function under physiological and pathophysiological conditions is of great scientific interest. Modulation of leukocyte/endothelium interaction is successfully used to treat multiple sclerosis patients with natalizumab, an antibody which prevents lymphocyte adhesion and thereby transmigration 6. However, various myopathies including the inflammatory subtypes still remain only poorly treatable to date. Therefore, elucidation of EC properties specifically in skeletal muscle endothelium could help to expand available treatments to myopathy or result in novel therapeutic approaches.
Immortalized endothelial cell lines (IECL) have been established and used for several in vitro models. These cell lines offer some advantages compared to primary microvascular EC due to immortalization and fast proliferation. Furthermore, primary cultured cells can undergo senescence and lose or change their morphology or physiological function after some time or passages. However, IECL only maintain some endothelial cell properties and cannot resemble the variety of different organ-borne primary microvascular EC. Of note, they are rarely derived from skeletal muscles. To date, only a single publication describes a human skeletal microvascular endothelial cell line named TSM15 5. In contrast, murine microvascular EC have not been described so far. Finally, primary cells from transgenic animals provide the opportunity to study genetic modifications in vitro. However, primary cell cultures also hold certain pitfalls. First, contamination with cells of no interest can interfere with the validity of experimental findings. Therefore, the accuracy of several selection steps and a high purity of isolated cells must be guaranteed. Cell suspension after dissociation of muscle tissue contains erythrocytes and different mononuclear cell types such as immune cells, satellite cells, pericytes, fibroblasts and endothelial cells 7. Therefore, cell fractions after the CD31 MCS step were tested for markers exclusively expressed by muscle satellite cells. As expected CD45– CD31– fraction and primary muscle cells showed Pax-7 and Cdh15 expression, whereas CD45– CD31+ and cultivated primary murine MMEC were negative for these markers after a second CD31 MCS step. Furthermore a quality control of isolated or cultured cells is inevitably to guarantee reliable and reproducible experiments. Several methods for quality control of primary murine MMEC are available: besides microscopy of morphological properties, flow cytometry, PCR and immunofluorescence stainings for characteristic EC markers (e.g. CD31, Sca-1) can be used. Purity assessment of freshly isolated primary murine MMEC was only about 60-70%, whereas a viability of about 70% could be detected. Microscopy of these cells partially shows cell conglomerates, which might consist of contaminating cells such as fibroblasts or pericytes attached to EC. After 7 days mainly flat and elongated cells and spheroid cells can be observed. However, two CD31 MCS passages resulted in high viability and purity. Alternative approaches to increase purity such as puromycin-containing media failed in primary murine MMEC while being effective in murine brain microvascular EC 8.
Since microvascular EC derived from capillaries express tight junction proteins, gene expression of Cld5, Ocln and Tjp1 in primary murine MMEC compared to differentiated muscle cells was examined. As expected all tight junction molecules were detected in primary murine MMEC. In comparison, muscle cells demonstrated only low expression of Tjp1, whereas no expression of Cld5 and Ocln could be determined. Similar expression patterns were previously demonstrated in whole murine skeletal muscle. Here, an expression on gene and protein level could be detected for Tjp1 but not for occludin 9. Further functional features such as transendothelial resistance could be measured.
The herein described protocol features only the isolation of microvascular EC from murine muscle tissue. However, it might be transferred to various other species as for example a similar protocol was published for rat microvascular EC derived from epididymal fat pads. Here, CD31 positive selection via MCS was preceded by density gradient centrifugation 10. Similar approaches were successful in macrovascular EC isolation from rat femoral arteries 11. A recently published method to isolate microvascular EC of heart and lung tissue uses CD31 and endoglin (CD105) as antigens for magnetic cell sorting 12. However, the purity of EC could not further be increased by using additional CD105 magnetic bead separation. Another possibility to isolate and purify primary murine MMEC is multicolor fluorescence activated cell sorting (FACS). A distinct population of Sca-1+, CD31+, CD34dim and CD45– cells from murine muscles could be isolated and was characterized as primary murine MMEC 13. An advantage of this method is a pure EC population which can be directly used or cultured for further experiments (Note that in our experience FACS leads to increased cell death due to higher cell stress). Further, dead cells or conglomerates of different cell types can significantly reduce the isolated cell numbers by FACS. Additionally, it is reported that primary murine MMEC isolated by FACS were unsuccessfully cultured at commonly used culture conditions of 21% O2 and 5% CO2. Here, the oxygen level had to be adjusted to 5% for a sufficient cultivation 13. Moreover, the FACS technique is more complex, time-consuming and the infrastructural prerequisites are expensive.
The musculus quadriceps femoris and the musculus triceps surae of male mice in the age of 4-12 weeks are most suitable for isolation of primary murine MEC as they are easily accessible and large enough for isolation of sufficient cell numbers (5 – 10 x 105 per gram muscle). Therefore, it must be ensured that conditions are comparable in independent experiments. As described above, primary cells can undergo senescence. Hence, use cells for respective experiments promptly in lower passages. After passage 8–10 primary murine MMEC change their morphology, demonstrate slow proliferation rates and lose their contact inhibition as signs for dedifferentiation and senescence.
Further there are some notes for troubleshooting for this protocol. Sterile working is essential to avoid contaminations. Quality controls are necessary to monitor purity and viability of isolated cells. Used materials in this protocol must be stored and applied according to manufacturer's instructions. Additional, age and sex of used animals, as well as different muscle groups might influence the quality and comparability of isolated cells in experiments.
The described protocol should be considered as platform method to isolate primary microvascular EC of skeletal muscles. Isolated cells can be used for several applications to gain further insights into blood-muscle-barrier function. Cells are suitable for both protein and RNA expression studies as well as functional assays (including transmigration and adhesion studies). However, this protocol was optimized for studies focusing on inflammatory processes and hence slight modifications might be necessary with respect to different research areas.
To model the complex spatial and functional cellular interactions within the MVU, primary murine MMEC can be cultivated in more complex 3D cell culture systems together with other cell types. Prospectively, it should also be possible to generate MVU organoids as it has been described for other tissues before 14. However, successful isolation of primary murine MMEC is inevitable for aiming at this next step.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by the "Else Kröner-Fresenius-Stiftung" (2018_A03 to TR), "Innovative Medizinische Forschung (IMF) Münster" (I-RU211811 to TR) and German Research Foundation (DFG, INST 2105/27-1, ME 3283/5-1, and ME 3283/6-1 to SGM). Illustrated images provided by Heike Blum.
Materials
| 0,25% Trypsin-EDTA | Thermo Fisher | 25200-056 | ready to use |
| ACK buffer | 150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA in water at a pH of 7.3 | ||
| Anti-mouse CD31-FITC (clone MEC13.3) | Biolegend | 102506 | Isotype control: FITC Rat IgG2a, κ Isotype Ctrl |
| Anti-mouse CD45-PE (clone 30-F11) | Biolegend | 103106 | Isotype control: PE Rat IgG2b, κ Isotype Ctrl |
| bFGF | Peprotech | 100-18B | Basic fibroblast growth factor |
| BSA | Sigma Aldrich | A4503 | |
| CD31 MicroBeads mouse | Miltenyi Biotec | 130-097-418 | |
| CD45 MicroBeads mouse | Miltenyi Biotec | 130-052-301 | |
| Collagenase-Dispase | Roche | 10269638001 | Collagenase from V. alginolyticus, Dispase from B. polymyxa |
| Corning Costar TC-Treated Multiple 6-Well Plates | Corning | 3516 | |
| Cy3-conjugated anti-rat IgG antibody | dianova | 712-166-153 | |
| DAPI (ProLong Gold antifade reagent with DAPI) | Thermo Fisher | P36935 | |
| Desoxyribonuclease | Sigma Aldrich | D4513 | Deoxyribonuclease I from bovine pancreas |
| Diethylpyrocarbonat treated water | Thermo Fisher | AM9916 | |
| DMEM, containing Glutamin Supplement and pyruvate | Thermo Fisher | 31966-021 | warm up to 37 °C before use |
| dNTP Mix (10 mM) | Thermo Fisher | R0192 | 1 mL |
| EDTA | Sigma Aldrich | E5134 | |
| FACS tubes | Sarstedt | 551,579 | |
| Falcon 70 μm Cell Strainer | Corning | 352350 | |
| FC buffer | 0.1% BSA, 0.2% NaN3, 2 mM EDTA | ||
| Fetal calf serum | Sigma Aldrich | F6178 | Fetal calf serum |
| Fixable Viability Dye eFluor780 | Thermo Fisher | 65-0865-14 | |
| Forceps (serrated, straight, 12 cm) | Fine Science Tools | 11002-12 | |
| Forceps (serrated, straight, 12 cm) | Fine Science Tools | 11009-13 | |
| Insulin syringe 100 Solo 1ml (Omnifix) | Braun | 9161708V | |
| large magnetiv columns (LS columns) | Miltenyi Biotec | 130-042-401 | for CD45-MACS-step |
| MCS buffer | 0.5% BSA, 2 mM EDTA in PBS at a pH of 7.2 | ||
| Medium magnetic column (MS column) | Miltenyi Biotec | 130-042-201 | for CD31-MACS-step |
| Nuclease free water | Thermo Fisher | R0581 | |
| PBS | Sigma Aldrich | Phosphate buffered saline, ready to use | |
| PCR buffer (5x) | Thermo Fisher | EP0742 | in a kit with the reverse transcriptase |
| Pecam1 rat α-mouse | SantaCruz | Sc-52713 | 100 µg/mL |
| Penicillin-Streptomycin | Sigma Aldrich | P4333 | |
| primary murine muscle cells | celprogen | 66066-01 | |
| Primer Cdh15 (M-Cadherin) | Thermo Fisher | Mm00483191_m1 | FAM labeled |
| Primer Cldn5 (claudin-5) | Thermo Fisher | Mm00727012_s1 | FAM labeled |
| Primer Ocln (occludin) | Thermo Fisher | Mm00500912_m1 | FAM labeled |
| Primer Pax-7 | Thermo Fisher | Mm01354484_m1 | FAM labeled |
| Primer Tjp-1 (Zonula occludens 1) | Thermo Fisher | Mm00493699_m1 | FAM labeled |
| Primer 18s rRNA (Eukaryotic endogenous control) | Thermo Fisher | 4310893E | VIC labeled |
| qPCR buffer (Maxima Probe/ROX qPCR Master Mix (2X) | Thermo Fisher | K0231 | 2 x 1,25 mL; for 200 reactions each |
| Random mixture of single-stranded primer | Thermo Fisher | SO142 | Random Hexamer Primer |
| Reverse Transcriptase (200 U/μL) + PCR buffer (5x) | Thermo Fisher | EP0742 | |
| Rnase Inhibitor (40 U/μL) | Thermo Fisher | EO0381 | |
| Scissor (cutting edge 23 mm, sharp/sharp) ) | Fine Science Tools | 14088-10 | |
| Scissor (cutting edge 42 mm, sharp/blunt) | Fine Science Tools | 14001-13 | |
| Speed Coating solution | PeloBiotech | PB-LU-000-0002-00 |
Referências
- Rodrigues, S. F., Granger, D. N. Blood cells and endothelial barrier function. Tissue Barriers. 3 (1-2), e978720 (2015).
- Michiels, C. Endothelial cell functions. Journal of Cellular Physiology. 196 (3), 430-443 (2003).
- Pierce, R. W., Giuliano, J. S., Pober, J. S. Endothelial cell function and dysfunction in critically ill children. Pediatrics. 140 (1), (2017).
- Nasdala, I., Wolburg-Buchholz, K., Wolburg, H., et al. A transmembrane tight junction protein selectively expressed on endothelial cells and platelets. Journal of Biological Chemistry. 277 (18), 16294-16303 (2002).
- Sano, H., Sano, Y., Ishiguchi, E., et al. Establishment of a new conditionally immortalized human skeletal muscle microvascular endothelial cell line. Journal of Cellular Physiology. 232 (12), 3286-3295 (2017).
- Kappos, L., Bates, D., Edan, G., et al. Natalizumab treatment for multiple sclerosis: Updated recommendations for patient selection and monitoring. Lancet Neurology. 10 (8), 745-758 (2011).
- Birbrair, A., Zhang, T., Wang, Z. M., et al. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Research. 10 (1), 67-84 (2013).
- Ruck, T., Bittner, S., Epping, L., Herrmann, A. M., Meuth, S. G. Isolation of primary murine brain microvascular endothelial cells. Journal of Visualized Experiments. (93), e52204 (2014).
- Hwang, I., An, B. S., Yang, H., Kang, H. S., Jung, E. M., Jeung, E. B. Tissue-specific expression of occludin, zona occludens-1, and junction adhesion molecule A in the duodenum, ileum, colon, kidney, liver, lung, brain, and skeletal muscle of C57BL mice. Journal of Physiology and Pharmacology. 64 (1), 11-18 (2013).
- Frye, C. A., Patrick, C. W. Isolation and culture of rat microvascular endothelial cells. In Vitro Cellular & Developmental Biology – Animal. 38 (4), 208-212 (2002).
- Le, G. Y., Essackjee, H. C., Ballard, H. J. Intracellular adenosine formation and release by freshly-isolated vascular endothelial cells from rat skeletal muscle: Effects of hypoxia and/or acidosis. Biochemical and Biophysical Research Communications. 450 (1), 93-98 (2014).
- Cheung, K. C., Marelli-Berg, F. M. Isolation of microvascular endothelial cells. Bio-protocol. 8 (12), (2018).
- Ieronimakis, N., Balasundaram, G., Reyes, M. Direct isolation, culture and transplant of mouse skeletal muscle derived endothelial cells with angiogenic potential. PLoS One. 3 (3), e0001753 (2008).
- Pham, M. T., Pollock, K. M., Rose, M. D., et al. Generation of human vascularized brain organoids. Neuroreport. 29 (7), 588-593 (2018).
