Assembling Molecular Shuttles Powered by Reversibly Attached Kinesins
Summary
We present a protocol to build molecular shuttles, where surface-adhered kinesin motor proteins propel dye-labelled microtubules. Weak interactions of the kinesins with the surface enables their reversible attachment to it. This creates a nanoscale system which exhibits dynamic assembly and disassembly of its components while retaining its functionality.
Abstract
This protocol describes how to create kinesin-powered molecular shuttles with a weak and reversible attachment of the kinesins to the surface. In contrast to previous protocols, in this system, microtubules recruit kinesin motor proteins from solution and place them on a surface. The kinesins will, in turn, facilitate the gliding of the microtubules along the surface before desorbing back into the bulk solution, thus being available to be recruited again. This continuous assembly and disassembly leads to striking dynamic behavior in the system, such as the formation of temporary kinesin trails by gliding microtubules.
Several experimental methods will be described throughout this experiment: UV-Vis spectrophotometry will be used to determine the concentration of stock solutions of reagents, coverslips will first be ozone and ultraviolet (UV) treated and then silanized before being mounted into flow cells, and total internal reflection fluorescence (TIRF) microscopy will be used to simultaneously image kinesin motors and microtubule filaments.
Introduction
The interactions governing the behavior of active nanosystems have always been characterized by long-lived, nearly irreversible bonds1,2,3,4,5,6,7,8. A well-studied example of this is the microtubule-kinesin system, where gliding microtubules are propelled by irreversibly surface-bound kinesin motors1,2,3,4,5. Systems in which the components are reversibly connected to one another have been studied theoretically9,10 and achieved at the macroscale11,12, but scaling these systems down to the nanoscale has been challenging. One of the major reasons for this is that breaking and reforming bonds between components often requires a large change in the environmental conditions. Even though such changes have been implemented in the past13,14,15, they would rely on modifying the system itself rather than adapting it to its environment. Designing molecular-scale systems in which components continually assemble and reorganize into structures without disturbing the overall environment in which the experiments take place will open the door to the exploration of a wide range of dynamic behaviors16,17.
Here, we describe and demonstrate the detailed protocol for creating a dynamically assembling and disassembling system functioning at the nanoscale. The system and its general behavior has been introduced earlier18: microtubule filaments are propelled by tracks of reversibly surface-bound kinesin-1 motors. These kinesin motor proteins are recruited from the solution to help propel microtubules forward, before desorbing again shortly afterwards. Once back in solution, they can be recruited again to propel a new microtubule. In the past13,14,15, the breaking and reforming of bonds required environmental modifications; in contrast, the environment of our flow cell remains unchanged while the kinesin motors interact with the surface.
This protocol will help interested researchers to (1) visualize all the steps of the protocol, and (2) assist with troubleshooting this type of assay. It has been derived from the procedures described in Howard et al. 199319.
Protocol
1. Solution preparation
CAUTION: Three of the reagents used in this protocol (toluene, dimethyldichlorosilane, and dithiothreitol) are highly toxic. Please consult the relevant material safety data sheets (MSDS) before use. Furthermore, parts of this protocol need to be performed under a higher level of protection, wearing safety glasses and two sets of protective gloves as indicated. Unless otherwise advised, the experiments can be performed on lab benches while using all the appropriate personal protective equipment (safety glasses, gloves, lab coat, full length pants, and closed-toe shoes).
NOTE: The concentrations of the ATP, kinesin, and microtubules that are used in this protocol can be modified given the needs of each experiment. However, if modified, please ensure that the final concentrations of the other reagents remain the same as given below. All experiments were performed at room temperature, (~25 °C).
- Preparation of stock solutions
NOTE: Prepare and aliquot the following solutions beforehand. All aliquots can be prepared at room temperature (~25 °C).- BRB80 buffer
NOTE: The buffer for most of the solutions used in this protocol is BRB80. BRB80 can be prepared in large quantities and stored in a -20 °C freezer.- Dissolve 24.2 g of piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) and 3.1 g of potassium hydroxide (KOH) in 800 mL in deionized water to make 800 mL of 100 mM PIPES. Add 100 mL of 10 mM magnesium chloride (MgCl2) and 100 mL of 10 mM ethylene glycol tetraacetic acid (EGTA) to get a final volume of 1 L. Raising the pH to 8 with potassium hydroxide (KOH) can help in the dissolution of PIPES and EGTA.
NOTE: The final concentrations of chemicals in the BRB80 buffer are 80 mM PIPES, 1 mM MgCl2, and 1 mM EGTA. - Adjust the pH of the buffer to 6.9 using KOH and hydrochloric acid (HCl).
- Dissolve 24.2 g of piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) and 3.1 g of potassium hydroxide (KOH) in 800 mL in deionized water to make 800 mL of 100 mM PIPES. Add 100 mL of 10 mM magnesium chloride (MgCl2) and 100 mL of 10 mM ethylene glycol tetraacetic acid (EGTA) to get a final volume of 1 L. Raising the pH to 8 with potassium hydroxide (KOH) can help in the dissolution of PIPES and EGTA.
- ATP
- Prepare a 1 mL solution of 100 mM ATP in ultrapure water. Pipet the solution into 10 μL aliquots. Store the aliquots in a -80 °C freezer.
- GTP
- Prepare a 500 μL solution of 25 mM GTP in ultrapure water. Pipet the solution into 5 μL aliquots. Store the aliquots in a -80 °C freezer.
- MgCl2
- Prepare a 1 mL solution of 100 mM MgCl2 in ultrapure water. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Casein
- Weigh 1 g of dry casein and transfer it to a 50 mL conical centrifuge tube. Add 35 mL of BRB80 buffer to the tube to dissolve the casein powder.
- Place the solution in a tumbler in a cold room to dissolve overnight. At this point, the solution will look thick and viscous.
- Leave the tube upright in a 4 °C fridge to allow for any large undissolved clumps to settle. Transfer the supernatant to a new tube.
- Spin the tube in a centrifuge at 1,000 x g to pellet out more precipitates. Once again, transfer the supernatant to a new tube.
- Repeatedly filter the solution using 0.2 μm (7 bar maximum pressure) filters. The solution being thick, many filters will get clogged, so repeat this step until the filter is clear and unclogged.
- Determine the casein concentration in the resulting solution using UV/Vis spectrophotometry, using casein’s extinction coefficient of 19 mM-1∙cm-1 at 280 nm20. Assuming a molecular weight of 23 kDa for casein, dilute the solution to a concentration of 20 mg∙mL-1 in BRB80.
- Pipet the solution into 20 μL aliquots and store the aliquots in a -20 °C freezer.
- D-Glucose
- Prepare a 1 mL solution of 2 M D-glucose in ultrapure water. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Glucose oxidase
- Prepare a 1 mL solution of 20 mg∙mL-1 glucose oxidase in BRB80. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Catalase
- Prepare a 1 mL solution of 0.8 mg∙mL-1 catalase in BRB80. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Dithiothreitol (DTT)
NOTE: Since DTT is slightly volatile and toxic, please perform the following steps under a fume hood.- Prepare a 1 mL solution of 1 M DTT diluted in ultrapure water. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Paclitaxel
- Prepare a 1 mL solution of 1 mM paclitaxel diluted in DMSO. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Dimethyl sulfoxide (DMSO)
NOTE: The DMSO used in these experiments is pure.- Pipet 1 mL of pure DMSO into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Creatine phosphate
- Prepare a 1 mL solution of 0.2 M creatine phosphate in ultrapure water. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Creatine phosphokinase
- Prepare a 1 mL solution of 200 units·L-1 creatine phosphokinase in ultrapure water. Pipet the solution into 10 μL aliquots. Store the aliquots in a -20 °C freezer.
- Nickel (II) sulfate
- Prepare 500 mL of a solution of 50 mM nickel (II) sulfate in ultrapure water. Store the solution at room temperature (~25 °C).
- Poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (PEG-PPG-PEG) solution
- Weigh out 2 mg of PEG-PPG-PEG (number average molecular weight: 14,600 g∙mol-1)-NTA powder on weighing paper. PEG-PPG-PEG-NTA is the triblock copolymer functionalized with a nitrilotriacetic acid (NTA) group. Refer to the Table of Materials for more details about the PEG-PPG-PEG-NTA.
- Transfer the powder into a 1.5 mL microcentrifuge tube. Add 1 mL of the stock nickel (II) sulfate solution to the tube. Vortex until powder is dissolved and no visible clumps remain.
- Store for up to one month at room temperature.
- BRB80 buffer
- Before starting an experiment
- Fill a bucket with ice.
- Take one aliquot from each of the 13 reagents described in sections 1.1.1 to 1.1.13 above and add them to the bucket, thus keeping them on ice. Thaw each of the reagents before use.
- Microtubule Preparation
NOTE: Microtubules were polymerized from a 20 μg aliquot of dye-labeled lyophilized tubulin. The excitation wavelength of the dye is of 647 nm.- Preparation of the microtubule growth buffer
- Pipet 21.8 μL of BRB80 buffer into a small (0.6 mL) microcentrifuge tube. Add 1 μL of the MgCl2 stock solution, 1 μL of the GTP stock solution, and 1.2 μL of the DMSO stock solution.
NOTE: Thus, the final concentrations of the reagents in the BRB80 buffer are 4 mM MgCl2, 1 mM GTP, and 5% (v/v) dimethyl sulfoxide.
- Pipet 21.8 μL of BRB80 buffer into a small (0.6 mL) microcentrifuge tube. Add 1 μL of the MgCl2 stock solution, 1 μL of the GTP stock solution, and 1.2 μL of the DMSO stock solution.
- Microtubule polymerization
- Add 6.25 μL of microtubule growth buffer directly into a 20 μg aliquot of labeled lyophilized tubulin. Vortex the aliquot for 5 s at 30 rps.
- Cool the aliquot on ice for 5 min before incubating it at 37 °C for 45 min and proceed to section 1.3.3.
- Microtubule stabilization
- Add 5 μL of the aliquoted paclitaxel solution to 490 μL of BRB80 buffer. Vortex the solution for 10 s at 30 rps.
- Once the 45 min of incubation for the microtubules are up, add 5 μL of the polymerized microtubule solution to the BRB80/paclitaxel solution.
NOTE: This 100-fold diluted, stabilized, microtubule solution will hereafter be referred to as MT100. It can be used for up to 5 days, and it can be diluted to obtain the desired microtubule density for each experiment.
- Preparation of the microtubule growth buffer
- Motility solution
- Enzymatic antifade, ATP regenerating system, and ATP
- Add 9.0 μL of the aliquoted casein solution to 291 μL of BRB80 buffer.
- If the desired ATP concentration for the experiment is lower than 1 mM, pipet 83 μL of the BRB80/casein solution into a new 0.6 mL microcentrifuge tube. Otherwise, pipet 85 μL of that solution into a new 0.6 mL microcentrifuge tube.
- Add 1 μL of D-glucose, 1 μL of glucose oxidase, 1 μL of catalase, and 1 μL of DTT to that tube.
NOTE: These chemicals constitute an enzymatic antifade cocktail21 that will reduce photobleaching by removing dissolved oxygen and quenching reactive radicals. This reduces photobleaching and microtubule disintegration caused by the excitation illumination during fluorescence imaging22,23. - For experiments in which the ATP concentration is chosen to be significantly lower than 1 mM, add the following reagents to the solution to create an ATP-regenerating system: 1 μL of the aliquoted creatine phosphate solution and 1 μL of the aliquoted creatine phosphokinase solution.
- Add 1 μL of the stock ATP solution to the motility solution. Flick or vortex the aliquot to homogenously distribute the chemicals.
NOTE: Thus, the final concentration of chemicals in the solution are 10 μM paclitaxel, 0.5 mg·mL−1 casein, 20 mM D-glucose, 200 μg·mL−1 glucose oxidase, 8 μg·mL−1 catalase, 10 mM dithiothreitol, 2 mM creatine phosphate (if added), 2 units·L−1 creatine phosphokinase (if added), and 1 mM ATP. This solution will hereafter be referred to as the motility solution.
- Kinesin
NOTE: The stock kinesin solution used in these experiments is prepared by G. Bachand at the Center for Integrated Nanotechnologies at Sandia National Laboratories and made available under a user agreement (https://cint.lanl.gov/becoming-user/call-for-proposals.php). The buffer used here consists of 40 mM imidazole, 300 mM NaCl, 0.76 g·L-1 EGTA, 37.2 mg·L-1 EDTA, 50 g·L-1 sucrose, 0.2 mM TCEP, and 50 μM Mg-ATP. For this series of experiments, the rkin430eGFP kinesin construct was used. This is a kinesin consisting of the first 430 amino acids of rat kinesin heavy chain fused to eGFP and a C-terminal His-tag at the tail domain24. It was expressed in Escherichia coli and purified using a Ni−NTA column. The concentration of the GFP-kinesin stock solution was 1.8 ± 0.3 μM as determined by UV/Vis spectrophotometry, using an extinction coefficient for GFP of 55 mM-1·cm-1 at 489 nm25, and taking into account that kinesin is a dimer.- Determine the desired kinesin concentration or surface density for the experiment. For typical assays, this concentration is 20 nM. If the kinesin concentration needs to be diluted more than a hundred-fold, dilute it in a solution of BRB80 that contains 0.5 mg·mL-1 of casein.
- Add 1 µL of kinesin solution to the motility solution in order to get a final concentration of approximatively 20 nM.
- Microtubules
- Add 10 μL of the MT100 solution prepared in section 1.3 to the motility solution.
NOTE: This motility solution can be used for up to 3 hours. After that time, the antifade system loses its effectiveness because the glucose in the solution is depleted by the enzymatic reaction.
- Add 10 μL of the MT100 solution prepared in section 1.3 to the motility solution.
- Enzymatic antifade, ATP regenerating system, and ATP
2. Assembling the flow cells
- Washing the coverslips
- For flow cells, use a large coverslip (dimension: 60 mm x 25 mm) and a small one (dimensions: 22 mm x 22 mm)19.
- Rinse all coverslips twice with ethanol and twice with ultrapure water. Sonicate the coverslips in ultrapure water for 5 min. Dry them in an oven at a temperature of 50–75 °C.
- Using a UV/ozone cleaner (see Table of Materials and manufacturer’s instructions), treat one side of each coverslip for 15 min. Perform this step can at room temperature (~25 °C) and in normal atmospheric condition (pressure of 1 atm).
- Carefully turn each coverslip to its other side (using tweezers) and UV/ozone treat that side as well.
- Sonicate the coverslips in ultrapure water again for 5 min before drying them again in the oven at a temperature of 50–75 °C.
- Treating the coverslips to enable PEG-PPG-PEG coating
NOTE: The dimethyldichlorosilane and the toluene used in this part of the protocol being highly toxic, perform the following steps under a fume hood, while taking the following precautions: wear two sets of protective gloves and a long-sleeved shirt under their lab coat. Tuck the edges of the gloves inside the sleeves of the shirt so that no skin from the forearms is directly exposed to the chemicals in case of a spillage. Wear protective glasses.- Dilute 25 mL of pure dimethyldichlorosilane in 475 mL of toluene. Immerse each coverslip in the dimethyldichlorosilane and toluene solution for 15 seconds.
- Wash the coverslips twice in toluene and three times in methanol. Dry the coverslips using pressurized nitrogen.
- Assembling coverslips into flow cells
- Once the coverslips are dry, cut a 2 cm x 2.5 cm piece of double-sided tape vertically into two 1 cm x 2.5 cm stripes.
- Put the large coverslip on a delicate task wiper and stick the tape stripes lengthwise along the edges of the coverslip to create a 1 cm x 2.5 cm area between the pieces of tape.
- Stick the small coverslip on top of the tape stripes to finish the flow cell assembly.
3. Flowing the solutions into a flow cell
- Flow approximatively 20 μL of the PEG-PPG-PEG solution into the assembled flow cell. The volume that is flown in the cell has to be large enough to fill the chamber. In the next steps, use the same volume when exchanging the solutions.
- Allow for the PEG-PPG-PEG solution to absorb on the surface for 5 minutes. Exchange the PEG-PPG-PEG solution with BRB80 buffer 3 times by flowing the buffer in. Flow the motility solution into the flow cell.
- Seal the edges of the flow cell with grease to prevent evaporation if the planned experiment is longer than an hour.
NOTE: The flow cell is now ready to be imaged.
4. Imaging a flow cell
- Perform the imaging using an objective-type total internal reflection fluorescence (TIRF) setup for both the microtubules and the kinesin motors (see Table of Materials).
NOTE: Here, we used a microscope with a 100x/1.49 numerical aperture objective lens, using two lasers, one with a wavelength of 642 nm and a maximum power of 140 mW, and another with wavelength of 488 nm and a maximum power of 150 mW. - Place a drop of immersion oil on the objective.
- Place the flow cell on the microscope platform and bring the objective up until there is contact between the oil on the objective and the flow cell.
- Use the microscope’s interlock cover system to block all laser light from escaping.
- Turn on the laser and focus on the lower surface of the flow cell. The microtubules are fluorescently labelled and excited at a wavelength of 647 nm. They will be imaged using a 642 nm laser while a 488 nm laser is used for the GFP-kinesin motors.
- Record the images or videos of interest. Typically, the laser power is of about 30 mW and an exposure time of 50 ms for both laser channels. Images can be recorded for as long as there is motility in the flow cell.
NOTE: The laser illumination is harmful for the naked eye and can cause irreparable damage. Please make sure that the illuminated area is completely covered with an opaque lid.
Representative Results
In these experiments, we used a 1,000 times dilution of the microtubules prepared in section 1.3.2. The kinesin concentration was 20 nM, and the ATP concentration was 1 mM. Imaging was performed using TIRF microscopy. Gliding microtubules were separately imaged from the kinesin motors: microtubules were visible upon excitation with a 647 nm laser (Figure 1, red), and the GFP-kinesin was visible when excited with a 488 nm laser (Figure 1, green). The time between excitation with the red and green light was less than 1 s. The time between frames was 10 s. Microtubules displayed stable gliding. The average microtubule gliding velocity was approximatively 800 nm/s. The microtubule surface density was 400 mm-2. Tracks of kinesin (green) appeared to extend beyond the trailing end of the microtubules (red) for several micrometers.
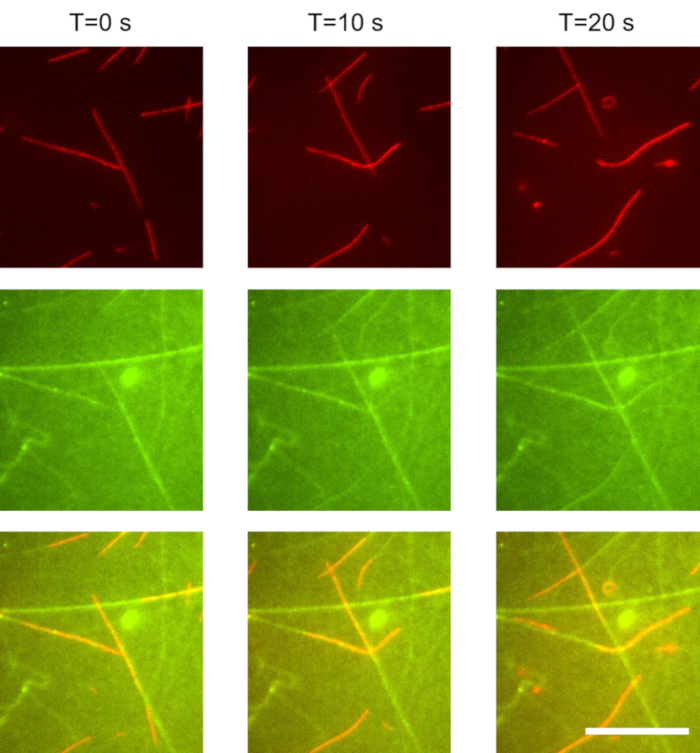
Figure 1: Gliding microtubules propelled by weakly surface-bound kinesin motors. As microtubules move forward, they accumulate kinesin motors from solution. These motors alternate between two states: single-bound to the microtubule, and double-bound to both the microtubule and the surface. When a double bound motor reaches the end of the microtubule, the motor is left behind and slowly desorbs from the surface with an off rate of approximately 0.1 s-1. As a result, trails of kinesin motors remain behind the microtubules. Top row: red (microtubule) channel. Middle row: green (kinesin) channel. Bottom row: combined red (microtubule) and green (GFP-kinesin) channel. Scale bar: 20 µm. Please click here to view a larger version of this figure.
Discussion
In this work, we present an active nanoscale system which self assembles weakly-binding building blocks to construct its own track. As shown in Figure 1, gliding microtubules accumulate kinesin motors from solution and deposit them on the surface. The kinesin motors remain in the wake of the microtubule for a short period of time before returning to solution. Thus, in this experiment, kinesin motors alternate between 3 states:
(1) A microtubule single-bound state: this is when a kinesin first binds to a microtubule. It exists in equilibrium with state (2).
(2) A double-bound state: in this case, a microtubule single-bound kinesin also binds to the surface via its His-tag. This double-bound state allows for microtubule propulsion.
(3) A single surface-bound state: a double-bound kinesin that has walked off the end of the microtubule and has not yet desorbed from the surface is in this state. These motors can be observed in Figure 1 (combined and green channels): they extend behind the tail of the microtubule for several micrometers and form its diminishing trail.
The most critical step of this protocol is the formation of the hydrophobic surface on the slide. Not only does it use dangerous chemicals, but it also allows the PEG-PPG-PEG functionalized with the NTA group to coat the surface, which then allows the kinesin to reversibly bind to the surface. Another important step is sealing the flow cell with grease. This allows for prolonged imaging without the liquid in the flow cell evaporating.
The primary modifications to this technique consist of changing microtubule concentration, kinesin concentration, and ATP concentration. Changing microtubule concentration will change the number of microtubules gliding on the surface. Changing kinesin concentration will change the number of kinesin molecules that can bind to the microtubule. However, increasing the kinesin concentration above the amounts already defined in this experiment could increase background fluorescence, making it more difficult to see the kinesin trails left behind gliding microtubules. Meanwhile, lowering ATP concentrations below 10 µM will significantly decrease microtubule gliding velocity. If this effect is desired, it is necessary to utilize an ATP regenerating system consisting of creatine phosphatase and phosphokinase.
A possible limitation of this technique is that, due to the large active kinesin content of the system, the ATP can be rapidly consumed, and experiments may last less than an hour in certain conditions. This would for example be the case if one used a twofold higher kinesin concentration and five-fold higher microtubule concentration than what is presented in this protocol.
In our previous work18, we studied the spatial distribution of kinesin motors along the microtubules, proving that gliding microtubules accumulate kinesin motors from solution, resulting in an increase of the density of motors along the length of the microtubule. We also found that the microtubules' gliding stability demonstrated a nonlinear dependence on the solution kinesin concentration and microtubule velocity.
The presented protocol paves the way for a more efficient use of protein motors in nanoscale engineered systems and for further investigation in the design of active nanosystems that are in dynamic equilibrium. Furthermore, the dynamic nature of this system allows it to serve as a model system for studying self-healing and dynamic replacement of molecular components, closing part of the gap between engineered and natural structures.
Declarações
The authors have nothing to disclose.
Acknowledgements
The authors gratefully acknowledge financial support under NSF grant NSF-DMR 1807514. The authors thank G. Bachand and V. Vandelinder for providing the GFP-kinesin protein. This work was performed, in part, at the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Los Alamos National Laboratory (contract no. DE- AC52-06NA25396) and Sandia National Laboratories (con- tract no. 97 DE-AC04-94AL85000). The authors thank Dr. Jennifer Neff and AllVivo Vascular for their gift of PEG-PPG-PEG functionalized with NTA.
Materials
| 488 nm laser | Omicron Laserage | LuxX 488-150 | |
| 642 nm laser | Omicron Laserage | LuxX 642 | |
| Casein | Sigma | C7078-500G | |
| Catalase from bovine liver | Sigma | C40-500MG | |
| Creatine Phosphate | Sigma | P-7936 | |
| Creatine Phosphokinase | Sigma | C3755-500UN | |
| D-Glucose | Sigma | G2133-50KU | |
| Dichlorodimethylsilane solution | Sigma | 40140-25ML | Toxic |
| Dimethyl Sulfoxide | Sigma | 34869-100ML | |
| Dithiothreitol | Sigma | D0632-5G | Toxic |
| Eclipse TI | Nikon Instruments | ||
| eGFP rkin430 | Provided by George Bachand | ||
| EGTA | Sigma | E4378-25G | |
| Falcon 50 mL Conical Centrifuge Tubes | Fisher Scientific | 14-959-49A | |
| Glucose Oxidase | Sigma | G0543-10KU | |
| Guanosine Triphosphate | Sigma | G8877-10MG | |
| Kimwipes Delicate Task Wipers | Sigma Pharmaceuticals | 8089 | |
| Magnesium Chloride | Sigma | M1028-100ML | |
| Methanol | Fisher Chemical | A412 | Toxic |
| Milli-Q Water Purification System | Millipore Corporation | ||
| Nickel Sulfate | Sigma | 656895-50G | |
| Paclitaxel | Sigma | T1912-5MG | |
| PIPES | Sigma | P-6757 | |
| Pluronic F108-NTA | Provided by Jennifer Neff and AllVivo Vascular | PEG-PPG-PEG-NTA | |
| Pluronic F-108 | Sigma | 542342-250G | PEG-PPG-PEG |
| Thermo Scientific Snap Cap Low Retention Microcentrifuge Tubes | Fisher Scientific | 21-403-190 | |
| Toluene | Fisher Chemical | T324 | Toxic |
| Tubulin, HiLyte647-labeled | Cytoskeleton, Inc. | TL670M | |
| UV Ozone Procleaner | BioForce Nanosciences | PC440 | |
| Whatman Puradisc syringe filters | Sigma | WHA67840402 | |
| Zyla 4.2 sCMOS Camera | Andor Technology | sCMOS 4.2 |
Referências
- Vale, R. D., Reese, T. S., Sheetz, M. P. Identification of a Novel Force-Generating Protein, Kinesin, Involved in Microtubule-Based Motility. Cell. 42, 39-50 (1985).
- Ray, S., Meyhofer, E., Milligan, R. A., Howard, J. Kinesin Follows the Microtubule’s Protofilament Axis. Journal of Cell Biology. 121, 1083-1093 (1993).
- Dennis, J. R., Howard, J., Vogel, V. Molecular shuttles: directed motion of microtubules along nanoscale kinesin tracks. Nanotechnology. 10, 232-236 (1999).
- Kawamura, R., Kakugo, A., Osada, Y., Gong, J. P. Microtubule bundle formation driven by ATP: the effect of concentrations of kinesin, streptavidin and microtubules. Nanotechnology. 21, 145603 (2010).
- Korten, T., Chaudhuri, S., Tavkin, E., Braun, M., Diez, S. Kinesin-1 Expressed in Insect Cells Improves Microtubule in Vitro Gliding Performance, Long-Term Stability and Guiding Efficiency in Nanostructures. IEEE Transactions on Nanobioscience. 15, 62-69 (2016).
- Whitesides, G. M., Grzybowski, B. Self-assembly at all scales. Science. 295, 2418-2421 (2002).
- Ringler, P., Schulz, G. E. Self-assembly of proteins into designed networks. Science. 302, 106-109 (2003).
- Boncheva, M., et al. Magnetic self-assembly of three-dimensional surfaces from planar sheets. Proceedings of the National Academy of Sciences of the United States of America. 102, 3924-3929 (2005).
- England, J. L. Dissipative adaptation in driven self-assembly. Nature Nanotechnology. 10, 919 (2015).
- Fialkowski, M., et al. Principles and Implementations of Dissipative (Dynamic) Self-Assembly. The Journal of Physical Chemistry B. 110, 2482-2496 (2006).
- Boncheva, M., Whitesides, G. M. Self-healing systems having a design stimulated by the vertebrate spine. Angewandte Chemie-International Edition. 42, 2644-2647 (2003).
- Rubenstein, M., Cornejo, A., Nagpal, R. Programmable self-assembly in a thousand-robot swarm. Science. 345, 795-799 (2014).
- Plaisted, T. A., Vakil Amirkhizi, A., Arbelaez, D., Nemat-Nasser, S. C., Nemat-Nasser, S. Self-healing structural composites with electromagnetic functionality. Proceedings of the Society of Photo-Optical Instrumentation Engineers. 5054, 372-381 (2003).
- Ghosh, S. K., Ghosh, S. K. . Self-Healing Materials. , 1-28 (2009).
- Burnworth, M., et al. Optically healable supramolecular polymers. Nature. 472, (2011).
- Bachand, G. D., Spoerke, E. D., Stevens, M. J. Microtubule-Based Nanomaterials: Exploiting Nature’s Dynamic Biopolymers. Biotechnology and Bioengineering. 112, 1065-1073 (2015).
- Gao, Y. W., Lei, F. M. Small scale effects on the mechanical behaviors of protein microtubules based on the nonlocal elasticity theory. Biochemical and Biophysical Research Communications. 387, 467-471 (2009).
- Lam, A. T. -. C., Tsitkov, S., Zhang, Y., Hess, H. Reversibly Bound Kinesin-1 Motor Proteins Propelling Microtubules Demonstrate Dynamic Recruitment of Active Building Blocks. Nano Letters. 18, 1530-1534 (2018).
- Howard, J., Hunt, A. J., Baek, S. Assay of microtubule movement driven by single kinesin molecules. Methods in Cell Biology. 39, 137-147 (1993).
- Huppertz, T., Fox, P. F., Kelly, A. L., Yada, R. Y. . Proteins in Food Processing (Second Edition). , 49-92 (2018).
- Wettermark, G., Borglund, E., Brolin, S. E. A regenerating system for studies of phosphoryl transfer from ATP. Analytical Biochemistry. 22, 211-218 (1968).
- Vigers, G. P. A., Coue, M., McIntosh, J. R. Fluorescent Microtubules Break Up Under Illumination. Journal of Cell Biology. 107, 1011-1024 (1988).
- Brunner, C., Hess, H., Ernst, K. -. H., Vogel, V. Lifetime of biomolecules in hybrid nanodevices. Nanotechnology. 15, S540-S548 (2004).
- Rogers, K. R., et al. KIF1D is a fast non-processive kinesin that demonstrates novel K-loop-dependent mechanochemistry. EMBO Journal. 20, 5101-5113 (2001).
- Patterson, G. H., Knobel, S. M., Sharif, W. D., Kain, S. R., Piston, D. W. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophysical Journal. 73, 2782-2790 (1997).
