There are five primary datasets providing useful information about key aspects of the procedure, the first being (1) efficiency of neural precursors transduction and co-transduction by lentiviral vectors. (2) An example of key features of the promoters employed to drive the "test gene". (3) An example of engineered cells ready for transplantation. (4) A cartoon including key procedural details of cell microinjection into the neonatal brain. (5) A synopsis of the whole morphometric pipeline.
As for (1), the capability of the prototypical lentiviral vectors delivered at different MOIs (2,4,8) to effectively transduce neocortical precursors was evaluated. In the first assay, neural precursors originating from early wild-type neocortices were infected by a lentiviral reporter, harboring the mCherry coding sequence (cds) under the control of the neuronogenic-lineage-specific pTα1 promoter, at MOI = 2, 4, 8, then allowed to differentiate. Co-immunoprofiling of their progenies for mCherry and the pan-neuronal marker Tubb3 showed that neurons were effectively transduced at frequencies of 64%, 69%, and 78%, respectively (Figure 1A). In a second assay, neocortical precursors were acutely coinfected by two lentiviral reporters, expressing EGFP and mCherry, under the control of the constitutive pPgk1 promoter, at MOIs = (2,2), (4,4), and (8,8). A few days later, co-immunoprofiling of their derivatives showed that the ratio between EGFP+mCherry+ and EGFP+mCherry± cells was 80%, 88%, and 93%, respectively (Figure 1B). These data suggest that, upon delivery of the engineering protocol envisaged for the present study, (a) the vast majority of neurons are transduced, and (b) almost all transduced neurons received the full lentiviral set needed for their characterization.
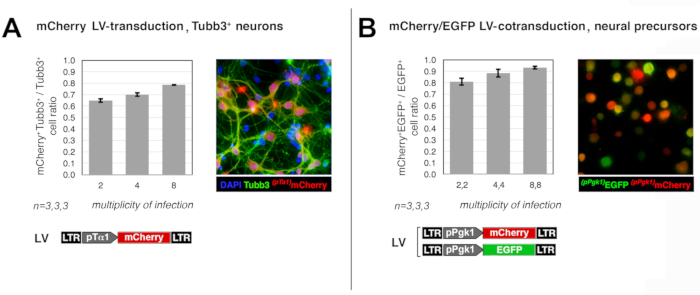
Figure 1: Efficiency of neural precursor cells transduction by lentiviral vectors. Panels report an evaluation of the efficiency by which E12.5 neocortical precursor cells are transduced (or co-transduced) upon delivery of dedicated lentiviral reporters at different multiplicities of infection (MOIs). In the former case (A), cells were acutely infected with the lentivirus (LV) pTα1-mCherry at MOI = 2, 4, 8, cultured in proliferative medium for 2 days, transferred to differentiative medium, and allowed to differentiate for 1 week. Upon fixation at day in vitro (DIV) 9, cultures were co-immunoprofiled for mCherry and the pan-neuronal marker Tubb3. mCherry and EGFP signals were detected by anti-RFP and anti-EGFP primary antibodies and revealed by Alexa-594- and Alexa-488-conjugated secondary antibodies, respectively. Finally, mCherry+Tubb3+/ mCherryTubb3+ ratios were calculated, averaged and plotted against the corresponding MOIs. In the latter case (B), cells were acutely infected with a 1:1 mix of two lentiviruses, encoding for the constitutively active pPgk-mCherry and pPgk-EGFP transgenes, at different MOIs (2, 4, 8 for each virus). Cells were cultured in proliferative medium for 4 days, trypsinized and, upon fixation, profiled for mCherry and EGFP immunofluorescence as above. Finally, mCherry+EGFP+/ mCherryEGFP+ ratios were calculated, averaged and plotted against the corresponding MOIs. Error bars represent S.E.M. Please click here to view a larger version of this figure.
As for (2), pTα1 and pSyn activity patterns can be inferred on the basis of expression profiles of fluoroprotein genes under their control. In this example, such control is direct in case of mCherry (Figure 2A), and indirect [i.e. mediated by a tetON second generation interface (Figure 2E)], in the case of EGFP (Figure 2B,C,D). Both promoters are specifically active within Tubb3+ postmitotic neurons (Figure 2B,C,D), but pTα1 also fires in a subset of Ki67+ intermitotic (neuronogenic) precursors (Figure 2A). Here, to provide an idea of limited variability of promoter activity when cells are engineered according to these standard conditions (Figure 2E), pTα1- and pSyn-driven EGFP expression levels within Tubb3+ neurons were quantified by Photoshop CS6 Histogram plugin. Then, raw data were normalized against medians and plotted against promoters (Figure 2F). Remarkably, single-cell fluorescence levels were densely clustered around medians: first and third quartiles equaled 0.86 and 1.16, as well as 0.89 and 1.12 in cases of pTα1 and pSyn, respectively.
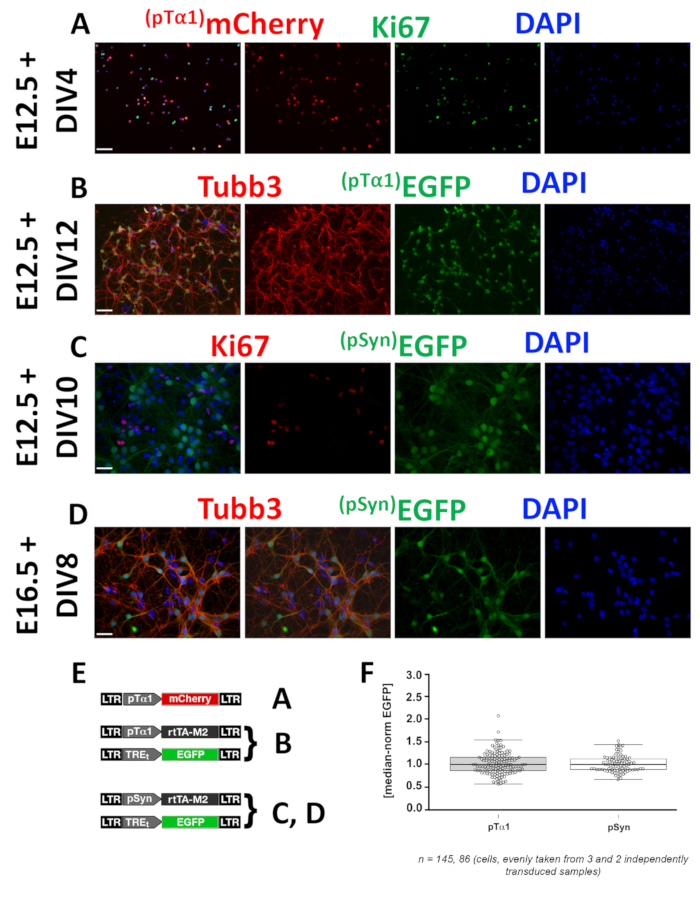
Figure 2: Profiling of neural precursor type-specific promoters suitable to drive GOI overexpression. Panels refer to characterization of Tubulin alpha 1 (pTα1) and Synapsin (pSyn) promoters, specifically active within the whole neuronal lineage and postmitotic neurons, respectively. Test were run in neocortical precursors harvested at different embryonic (En) ages, acutely infected with dedicated lentiviral mixes [pTα1-mCherry in (A); pTα1-rtTA, TREt-EGFP in (B); pSyn-rtTA, TREt-EGFP in (C,D); 2µg/ml doxy ab initio], and cultured in proliferative medium (A), proliferative (2 days) followed by differentiative medium (B,C), or differentiative (D) medium. Upon fixation at day in vitro (DIV) n, postmitotic neurons were identified by αTubb3 and intermitotic precursors by αKi67 immunofluorescence; mCherry and EGFP were detected as shown in Figure 1. Signals were revealed as shown in Figure 1. Scale bars: 100 µm in (A,B) and 50 µm in (C,D). Shown are (E) dedicated lentiviral mixes used in this study with references to the figure panels. Shown is a (F) a scatter plot of pTα1- and pSyn-driven EGFP signal in Tubb3+ cells referred to in (B) and (D), respectively. Boxed are cells falling between 25th and 75th percentiles; whiskers refer to 10th and 90th percentiles. Please click here to view a larger version of this figure.
As for (3), 3 days after lentiviral transduction, MtaptEGFP/+ "green" neurospheres express the Pgk1p promoter-driven, mCherry red fluorescent protein ("control" spheres) or not ("test" spheres) (Figure 3A,B). All spheres are dissociated to single cells, ensuring there are no clumps lefts, and the corresponding suspensions are mixed 1:1. The resulting mix (Figure 3C) is placed on ice and used within 20 min for transplantation.
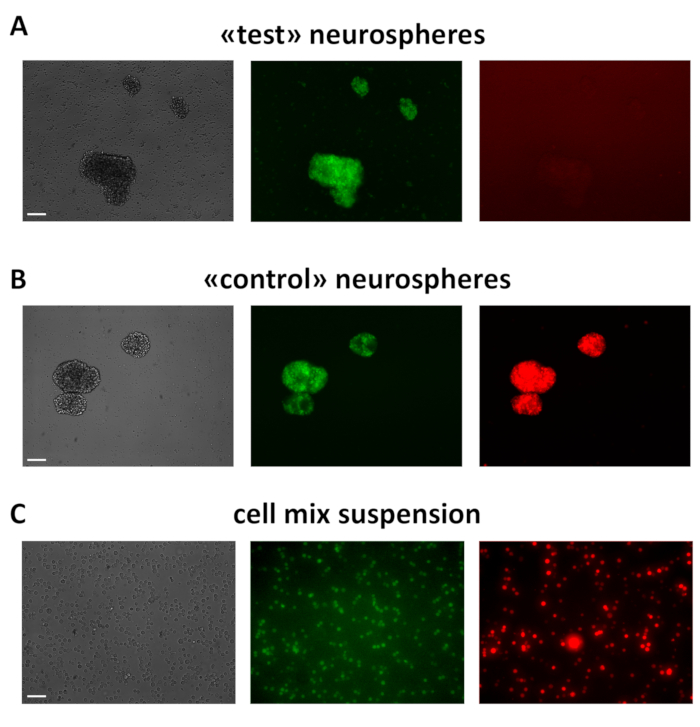
Figure 3: Example of test ("only green") and control ("green-red") DIV2 neurospheres and cell suspension ready for transplantation. (A,B) These spheres were obtained from MtaptEGFP/+E 12.5 neocortical precursors, acutely infected by lentiviral mixes "pCAG_lacZ, Pgk1p_tTA, TREt_GOI" (A) and "Pgk1p_mCherry, Pgk1p_tTA, TREt_PLAP" (B), respectively, each LV at MOI = 8, and kept in proliferative medium. (C) Cell suspension was obtained by dissociating "test" and "control" neurospheres (A,B) to single cells and mixing them 1:1. Scale bars: 200 µm in (A,B) and 100 µm in (C). Please click here to view a larger version of this figure.
As for (4), the procedure for cell injection into the neonatal brain includes three key steps. The capillary, placed on the mid-parasagittal plane, is entered into the lateral ventricular cavity via the frontal cortical wall (Figure 4A). Next, to prevent damage of the ganglionic eminence, the capillary is rotated 30° within the horizontal plane, so that the tip points medially/caudally (Figure 4B). Last, the cell suspension is delicately ejected into the lateral ventricular cavity, where it forms an easily distinguishable, light-blue "cloud" (Figure 4C).
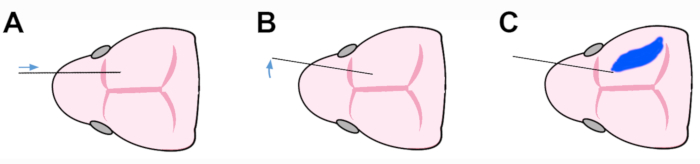
Figure 4: Schematics of the three steps procedure employed for cell injection into the neonatal brain. (A) The capillary is entered into the lateral ventricle through the frontal neocortical wall. Then, (B) it is rotated medialward, to prevent damage of ganglionic eminence. Finally (C), the cell suspension is gently injected into the lateral ventricle, where it forms a light blue cloud. Please click here to view a larger version of this figure.
As for (5), the analytical procedure includes four main steps. First, optical confocal sections of transplanted neuron-rich fields are flattened, according to MAX Z-projection modality so generating RGB .tiff files. Here, as an example, only "green test" neurons and "yellow control" neurons, originating from co-transplanted precursors, can be easily distinguished (it should be noted that mCherry may alternatively be used to label "test" neurons) (Figure 5A). Then, an idealized, skeletonized camera lucida version of the picture is generated by a suitable graphic software (see step 4.2.6) (Figure 5B) by an operator blind of red channel. Blindness of red channel is of paramount importance in order to prevent any unintentional bias in data evaluation. Next, single-neuronal silhouettes are fed into NeurphologyJ software as black-and-white pictures and values of the three primary parameters "total number of exitpoints" (attachment_points), "total number of endpoints" (endpoints) and "total neurite length" (neurite_length) are collected (Figure 5C). Finally, secondary parameters of each neuron are calculated, averaged and statistically evaluated by Excel software (Figure 5D).
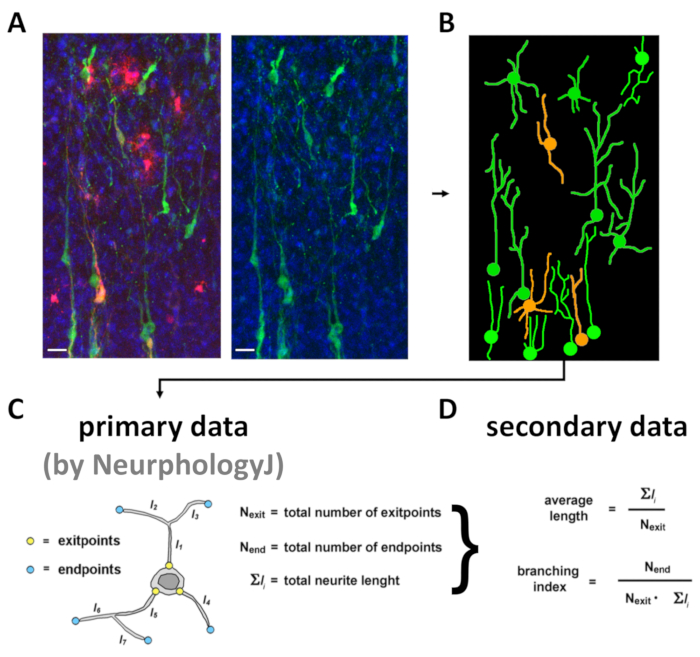
Figure 5: Representative flowchart of morphometric analysis of transplanted brains. Upper row panels show: (A) a MAX z-projection .tiff image of medial neocortex, fixed 10 days after engineered cell transplantation (green = Mtapt-driven, neuron-restricted EGFP; red = mCherry, constitutively labeling "control" cells; blue = DAPI), the green channel signal, and (B) its skeletonized e-camera lucida rendering. Scale bars: 50 µm. The bottom row includes: (C) primary morphometric parameters extracted from neuronal skeletons by NeurphologyJ software analysis (neurite_length as Σli, attachment_points as Nexit and endpoints as Nend) and (D) secondary parameters calculated on their basis. This figure has been modified from Chiola et al.21. Please click here to view a larger version of this figure.
