In cultured hippocampal neurons, TLCN was abundantly localized to the dendritic filopodia, shaft, and soma and colocalized with F-actin (Figure 1A, B). When polystyrene microbeads were added to cultured hippocampal neurons, the beads were automatically coated with vitronectin (VN) derived from fetal bovine serum (FBS) in the culture medium; they were mainly bound to dendrites, and they induced the formation of phagocytic cups (Figure 1B-E). Phagocytic cups were based on sheet-shaped actin filaments around microbeads on dendrites. TLCN, phospho-ERM, and PI(4,5)P2, which are markers for dendritic filopodia, are highly accumulated around beads (Figure 1D)8,15. Phagocytic cups were only formed on wild-type hippocampal neurons, but not on TLCN-deficient hippocampal neurons (Figure 2A-D). Thus, the phagocytic cup formation was crucially dependent on the presence of TLCN in dendrites.
Constituents of the dendritic filopodia appeared similar to those of phagocytic cups. Next, we purified phagocytic cups instead of dendritic filopodia. The protocol for the purification of phagocytic cups is schematically shown in Figure 3. Magnetic microbeads were added to wild-type cultured hippocampal neurons, which induces the formation of phagocytic cup structures. The phagocytic cup structures were lysed with a weak detergent. The microbeads were collected after the lysis using a magnet separator. After washing of the beads, their binding proteins were boiled and eluted in an SDS sample buffer.
The amount of proteins in the bound and unbound fraction was measured using the BCA protein assay kit. The same amount of proteins in the unbound and bound fractions was separated by SDS-PAGE and stained by silver staining (Figure 4A). The protein band patterns were almost the same for the unbound and bound fractions, but the intensities at 50 and 70 kDa in the bound fraction were lower than those in the unbound fraction. However, the band intensity was not obviously different between the unbound and bound fractions prepared from TLCN-deficient culture hippocampal neurons. To confirm the purification of phagocytic cup structures, we performed Western blotting using anti-TLCN-C, anti-bovine vitronectin, anti-actin, and anti-α-tubulin (Figure 4B). TLCN and VN were mainly detected in the bound fraction. Actin, ezrin, Gαq, PLCβ1, MAP-2, and spectrin were detected in both the bound and unbound fractions. Moesin, PSD-95, α-actinin, and α-tubulin were detected in the unbound fraction.
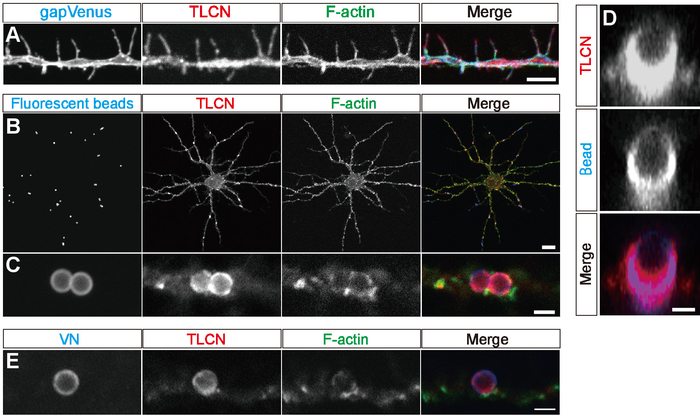
Figure 1: Localization of TLCN and F-actin in dendritic filopodia and phagocytic cups. (A) Immunofluorescence staining of dendrites of a cultured hippocampal neuron expressing gapVenus with anti-GFP antibody (blue in a merged image), anti-TLCN antibody (red in a merged image), and phalloidin (green in a merged image). TLCN and F-actin are abundantly observed in dendritic filopodia. (B, C) Formation of phagocytic cup structures on neuronal dendrites. Fluorescent microbeads (blue in merged images of B, C) added to cultured hippocampal neurons strongly adhere onto dendrites and induce the accumulation of TLCN (red in merged images of B, C) and F-actin (green in merged images of B, C). (D) A lateral view of a dendritic phagocytic cup reconstructed from confocal images reveals TLCN (red in merged images of D) surrounding the fluorescent bead (blue in merged images of D). (E) A dendritic phagocytic cup induced by a magnetic microbead attached onto a neuronal dendrite and immunostained with anti-VN antibody (blue in merged images of E), anti-TLCN antibody (red in merged images of E), and phalloidin (green in merged images of E). Scale bars = 1 μm in (D); 2 μm in (A), (C), and (E); and 20 μm in (B). This figure has been modified from a previous study18. Please click here to view a larger version of this figure.
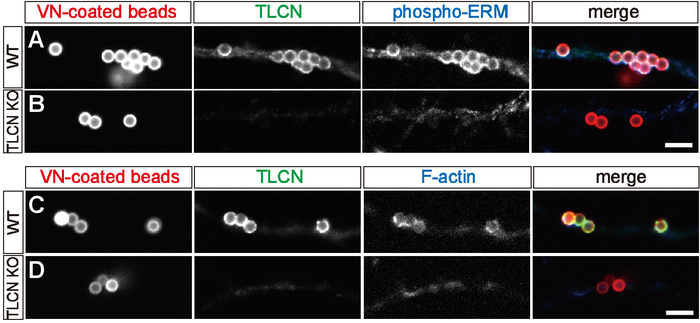
Figure 2: TLCN-dependent formation of phagocytic cup-like structure. (A–D) Triple fluorescence images of wild-type (WT; A, C) and TLCN-deficient (KO; C, D) neurons treated with VN-coated fluorescent beads (red in merged images of A, B, C, D) and labeled with anti-TLCN antibody (green in merged images of A, B, C, D) and anti-phospho-ERM antibody (blue in merged images of A, B) or Alexa488-phalloidin (blue in merged images of C, D). Scale bars = 5 μm. This figure has been modified from a previous study15. Please click here to view a larger version of this figure.
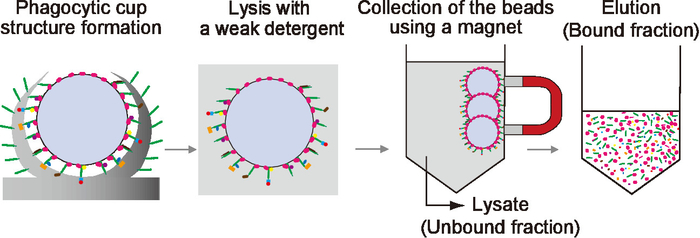
Figure 3: A schematic diagram illustrating the purification procedure of the dendritic filopodia-rich fraction. Magnetic microbeads were added onto cultured hippocampal neurons to induce the formation of dendritic phagocytic cups. After 1 day of incubation, the neurons were solubilized with lysis buffer containing 0.01% Triton X-100. The beads were separated from the unbound fraction using a magnetic separator. After washing, the bound proteins were eluted with SDS sample buffer and used as the bound fraction. Red: VN, green: TLCN, other colors: bound proteins. This figure has been modified from a previous study18. Please click here to view a larger version of this figure.
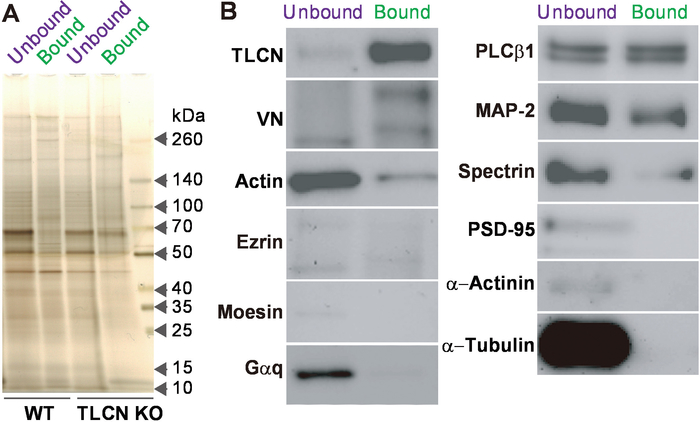
Figure 4: Confirmation of phagocytic cup fraction. (A) Silver staining of proteins in the unbound and bound fractions of the microbeads. The same amount (50 ng) of proteins in the unbound and bound fractions purified from wild-type (WT) and TLCN-deficient (TLCN KO) hippocampal neurons were separated by SDS-PAGE and visualized with silver staining. (B) Western blot analysis of the unbound and bound fractions. The same amount (50 ng) of proteins were separated by SDS-PAGE and subjected to Western blot analysis using anti-TLCN, anti-VN, anti-actin, anti-ezrin, anti-moesin, anti-Gαq, anti-PLCβ1, anti-MAP-2, anti-spectrin, anti-PSD-95, anti-α-actinin, and anti-α-tubulin antibodies. Note that TLCN, VN, actin, ezrin, PLCβ1, MAP-2, and spectrin are observed in the dendritic filopodia-rich fraction. This figure has been modified from a previous study18. Please click here to view a larger version of this figure.
