All animal handling and surgical procedures were performed by a protocol approved by the Augusta University Institutional Animal Care and Use Committee (IACUC). Mice were fed standard diet and water ad libitum.
1. Isolation of primary mouse fibroblasts from adult Dmdmdx mice
- Euthanize adult Dmdmdx mice (male, 2 months old) by CO2 asphyxiation and thoracotomy according to IACUC approved by Medical College of Georgia, Augusta University. Cut the tail with a sterile scalpel in a sterile condition under the laminar hood. Rinse the tail with 70% ethanol for 5 min, and then wash with sterile phosphate-buffered saline (PBS) in a 6 cm dish.
- Peel the tail skin off by a sharp incision from the base to the tail tip along the tail skin, and then gently peel off the tail skin with tweezers. Mince the skin to a size of 1 mm3 using a sterile scalpel and move the minced skin tissue to a 6 cm dish in Dulbecco’s minimal essential medium/Ham’s F12 (DMEM/F12) containing 0.1% collagenase IV and 1 U/mL of dispase.
- Digest the skin tissue in a culture dish for 2 h at 37 °C in a 5% CO2 incubator.
- Coat a 6-well plate with fibronectin and 0.2% gelatin (1 mL of fibronectin in 199 mL of 0.2% gelatin; Table of Materials) and incubate at 37 °C for 1 h.
- Dissociate the digested skin tissue with a 1 mL pipet tip and transfer the tissue and supernatant to a sterile 15 mL conical centrifuge tube. Centrifuge at 217 x g for 3 min at room temperature, discard the supernatant and resuspend the pellet in 1.5 mL of fibroblast medium (Table of Materials).
- Culture the pellets including incomplete digested skin tissue from step 1.5 on the 6-well plate coated with fibronectin and 0.2% gelatin from step 1.4 in a 37 °C, 5% CO2 incubator. Replace the medium 24 h after the initial plating to remove unattached cells, and change the medium every 48 h.
2. Reprogramming mouse skin fibroblasts into iPSCs
- Two days before transduction, digest mouse dermal fibroblasts from step 1.6 with the cell detachment solution (Table of Materials) in a 37 °C incubator with a humidified atmosphere of 5% CO2 for 5 min.
- Count cells using a hemacytometer and centrifuge at 217 x g for 3 min.
- Seed cells at a density of 1−2 x 105 cells per well onto a 6-well plate and culture with fibroblast medium (Table of Materials) in a 37 °C incubator with a humidified atmosphere of 5% CO2.
- On the day of transduction (day 0), estimate the cells and calculate the volume of each virus required to reach the target multiplicity of infection (MOI) of 5, 5, and 3 (i.e., KOS MOI = 5, hc-Myc MOI = 5, hKlf4 MOI = 3) according to the commercial manual.
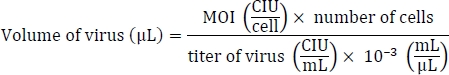
- Thaw three Sendai tubes in a 37 °C water bath for 5−10 s and add the calculated volumes of each of the three Sendai tubes to 1 mL of fibroblast medium (Table of Materials).
- Remove the fibroblast medium from step 2.3 and add the reprogramming virus mixture to the wells containing the cells. Incubate the cells overnight in a 37 °C incubator with a humidified atmosphere of 5% CO2.
- Replace the medium with fresh fibroblast medium 24 h after transduction. Culture the cells for one week with medium exchange every other day.
- Harvest infected mouse fibroblasts on day 7 after transduction with 0.05% trypsin/EDTA and place on dishes that are previously coated with fibronectin and 0.2% gelatin.
- Culture the infected mouse fibroblasts from step 2.6 with complete mouse embryonic stem cell (ES) growth medium (Table of Materials) in a 37 °C incubator with a humidified atmosphere of 5% CO2 and change medium daily.
- From the 8th day, observe the plate under an inverted microscope every other day to identify the appearance of cell clumps with the morphology of mouse ES.
3. Using alkaline phosphatase live stain and flow cytometry to quantify reprogramming efficiency
- Remove the culture media from each well and rinse with DMEM/F-12 for 2−3 min.
- Apply 2 mL of 1x alkaline phosphatase (AP) live stain working solution (1:500 dilution in DMEM/F-12) to the adherent cells, and incubate in a 37 °C incubator with a humidified atmosphere of 5% CO2 for 30 min.
- Aspirate the AP live stain and wash twice with PBS for 5 min each.
- Digest the cells with the cell detachment solution (Table of Materials) in a 37 °C incubator with a humidified atmosphere of 5% CO2 for 5 min. Perform flow cytometry to determine the reprogramming efficiency.
4. Selecting and harvesting ES-like cells
- Examine the colonies from step 2.8 under an inverted microscope.
- Mark the colonies at the bottom of the dish with a self-inking object marker.
- Apply greased cloning rings to cover the marked cell colonies. Add 100 μL of 0.05% trypsin/EDTA to each cloning ring at 37 °C for 5 min, and then transfer the digested cells with 100 μL pipette tips to 48-well culture plates containing mES growth medium.
- Incubate the cells in 48-well culture plates in a 37 °C incubator with a humidified atmosphere of 5% CO2. Passage the cells to 6 cm dish when they reach 70% confluence.
- Repeat steps 4.3 and 4.4 for several times until uniform dorm-shaped clones are obtained.
5. Freezing iPSCs for cryopreservation
- Dissociate the selected iPSCs from step 4.5 with trypsin, versene, and chick plasma (TVP; Table of Materials) solution at 37 °C in a 5% CO2 incubator for 30 min.
- Collect the cells in a sterile 15 mL conical tube and centrifuge at 217 x g for 3 min at room temperature.
- Aspirate the supernatant and re-suspend the cell pellet in 2 mL of mouse ES frozen medium (Table of Materials) to obtain 1 mL per cryovial.
- Add cells to cryovials and freeze using a freezer container that provides the critical, repeated -1 °C/min cooling rate required for cryopreservation at -80 °C overnight.
- Transfer frozen vials into a liquid nitrogen tank.
6. Immunofluorescence staining for stem cell markers in iPSCs
- Seed iPSCs from step 5.1 cultured with mES medium into an 8-well chamber slide coated with poly-D-lysine/laminin (Table of Materials) at the appropriate density to achieve between 1−2 x 104 cells per well and incubate in a 37 °C incubator with a humidified atmosphere of 5% CO2 for 48 h.
- Immerse the slides in 4% formaldehyde for 15 min at room temperature, and then immerse the slides in PBS twice for 5 min each time.
- Incubate sections with a mouse IgG-blocking reagent (Table of Materials), and 5% goat serum for 1 h at room temperature.
- Dilute the primary antibodies in protein diluent (mouse-anti-SSEA1, 1:100, rabbit-anti-Nanog, 1:500; rabbit-anti-POU class 5 homeobox 1 [OCT4], 1:500; rabbit-anti-SRY-box 2 [SOX2], 1:500; rabbit-anti-Lin-28 homolog A [Lin28A], 1:400) (Table of Materials). Apply antibodies to the cells and incubate at 4 °C overnight in a humidified chamber.
- Discard the primary antibody solution and wash the slides 3x in PBS.
- Apply second antibodies (Alexa488-conjugated goat-anti-mouse antibody and Alexa555-conjugated goat-anti-rabbit, 1:400 each) in M.O.M. protein diluent on slides and incubate for 45 min at room temperature.
- Wash the slides 3x in PBS and mount sections with mounting medium containing 4’6-diamidino-2-phenylindole (DAPI).
- Take pictures with a confocal microscope.
7. Investigating the pluripotency of iPSCs in vivo
- Dissociate the iPSCs from step 5.1 into single cells using TVP solution in a 37 °C incubator with a humidified atmosphere of 5% CO2 for 30 min.
- Count cells using a hemacytometer and centrifuge at 217 x g for 3 min.
- Aspirate the supernatant and resuspend the pellet with mES medium in a 1.5 mL sterile centrifuge tube at a concentration of 5 x 105 cells/30 μL for cell transplantation.
- Remove hair from both hind limbs of immunodeficient mice using hair clippers.
- Anesthetize mice with ketamine (100 mg/kg) and clean the injection site with 75% alcohol.
- Inject 30 μL of iPSC suspension from step 7.3 intramuscularly into the gastrocnemii, using a 31 G needle.
- Two weeks after the injection, harvest the mouse gastrocnemii and embed in optimal cutting temperature (OCT) compound, snap freeze and cut into 5-μm sections15,16.
- Fix sections in 4% formaldehyde for 15 min at room temperature, and then wash the slides twice in PBS for 5 min each time.
- Block the cells with 5% goat serum protein diluent for 1 h at room temperature.
- Add diluted primary antibody (rabbit anti-AFP, 1:50; rabbit anti-SMA, 1:50; rabbit anti-TH, 1:50) to the slides and incubate at 4 °C overnight in a humidified chamber.
- Discard the primary antibody solution, wash the cells 3x (5 min/wash) in PBS, add a 1:400 diluted Alexa555-conjugated goat-anti-rabbit antibody to slides, and incubate for 45 min at room temperature.
- Wash slides 3x (5 min/wash) in PBS, and mount sections with mounting medium containing DAPI.
8. Construction of CRISPR/Cas9 lentiviral vector targeting introns flanking dystrophin exon 23
- Design two pairs of gRNA oligos targeting intron-flanked dystrophin exon 23 via http://crispor.tefor.net/crispor.py.
NOTE: The designed pairs are as follows:
i22sense: 5’-CACCGTTAAGCTTAGGTAAAATCAA– 3’
i22anti-sense: 5’-AAACTTGATTTTACCTAAGCTTAAC-3’
i23sense: 5’-CACCGAGTAATGTGTCATACCTTCT– 3’
i23anti-sense: 5’-AAACAGAAGGTATGACACATTACTC-3’). - Digest and dephosphorylate 5 µg of lentiviral CRISPR plasmid (lenti-CRISPRv2-blast [a gift from Mohan Babu] and lenti-Guide-Hygro-iRFP670 [a gift from Kristen Brennand]) (Table of Materials) with BsmB1/Esp3I for 30 min at 37 °C. For 5 µg of plasmids, add 3 µL of BsmB1 restrict enzyme, 3 µL of fast alkaline phosphatase, 6 µL of 10x enzyme digest buffer and 0.6 µL of 100 mM DTT in 60 µL reaction).
- Load the reactions onto a 0.8% agarose gel. Run the gel at 100−150 V for 30 min.
- Purify digested plasmid in-gel using a gel extraction kit (Table of Materials) and elute in 20 µL of H2O.
- Phosphorylate and anneal each pair of the gRNA oligonucleotides containing 1 µL of each oligonucleotide at 100 µM, 1 µL of 10x T4 ligation buffer, 0.5 µL of T4 polynucleotide kinase (PNK), 6.5 µL of ddH2O at 37 °C for 30 min, and then 95 °C for 5 min and then ramp down to 25 °C at 5 °C/min.
- Ligate a 1:200 dilution of the annealed gRNA oligonucleotides into the plentiCRISPR V2-Blast or plentiGuide-Hygro-iRFP670. Mix 50 ng of BsmB1/Esp3I digested vectors with 1 µL of diluted oligo duplex and 5 µL of 2x ligase buffer plus 1 µL of ligase in a 11 µL reaction system and incubate for 10 min at room temperature.
- Perform transformation with 3 µL of ligation product into 50 µL competent cells (Table of Materials) according to the manufacturer’s instructions.
- Spread the transformed competent cells in an agar plate with 100 µg/mL carbenicillin and incubate at 31.5 °C for 18 h.
- Pick colonies with 10 µL sterile pipette tips and culture in 5 mL of terrific broth (Table of Materials) containing 100 µg/mL carbenicillin at 31.5 °C, 185 rpm in a shaker incubator for 21 h.
- Purify the plasmid DNA using the mini-prep and midi-prep kits (Table of Materials).
- Verify the mini-prep plasmids by restriction digestion. For the 20 µL reaction system, add 1 µg of plasmid DNA, 2 µL of digest reaction buffer (Table of Materials) and 1 µL of restriction enzyme mixture (0.5 µL of KpnI-HF and 0.5 µL of AgeI-HF for plentiCRISPR V2-Blast-i22; 0.5 µL of NotI-HF and 0.5 µL of EcoRI-HF for plentiGuide-Hygro- iRFP670- i23). Incubate the reaction system for 1 h at 37 °C.
- Load the reactions onto a 0.8% agarose gel. Run the gel at 100−150 V for 30 min.
NOTE: The correct bands for plentiCRISPR V2-Blast-i22 should be 622 bp and 12.2 kb, and the correct bands for plentiGuide-Hygro- iRFP670- i23 should be 2.6 kb and 7.1 kb.
9. Lentiviral vector packaging
- Culture 7 x 105 293FT cells in 5 mL of DMEM media containing 10% fetal bovine serum in a 6 cm dish overnight at 37 °C, 5% CO2.
- Prepare a cocktail (1 µg of plentiCRISPR V2-Blast-i22 or plentiGuide-Hygro-iRFP670-i23, 750 ng of psPAX2 packaging plasmid, 250 ng of pMD2.G envelope plasmid, and 5 µL of transfection reagent A [Table of Materials] in 100 µL of reduced serum MEM media).
- Prepare a mixture of 5 µL of transfection reagent B (Table of Materials) in 100 µL of reduced serum MEM media.
- Add 100 µL of transfection reagent B mixture to plasmid mixture from step 9.2 and incubate for 5 min at room temperature.
- Add the DNA-lipid complex (from step 9.4) dropwise to the 293FT cells. Incubate overnight at 37 °C with 5% CO2.
- Add virus production enhancer (500x) (Table of Materials) to each dish the next day and incubate for 24 h at 37 °C, 5% CO2.
- Collect medium from cells using pipettes on the next two days and filter the medium through a 0.45 µm filter to remove the cells.
10. Concentration and purification of lentiviral vectors
- Precipitate lentiviral vector in the medium of step 9.7 overnight at 4 °C with 5x polyethylene glycol 4000 (PEG4000, 8.5% final concentration) and 4 M NaCl (0.4 M final concentration).
- Centrifuge the viral media containing the PEG4000 solution at 2,095 x g and 4 °C for 30 min, remove and discard the supernatant.
- Resuspend the pellets with 500 µL of serum reduced MEM media (lentivirus titer: lenti-CRISPR V2-gRNAi22: 1.56 x 108, lenti-iRFP670-gRNAi23: 1.3 x 108, lenti-CRISPR V2-control: 3.13 x 107, lenti-iRFP670-control: 5.9 x 107). Store at -80 °C until use.
11. Deletion of exon 23 in mouse iPSCs with two guide RNAs (gRNAs) coupled with Cas9
- Plate the mouse iPSCs from step 4.5 in a 24-well plate coated with fibronectin and gelatin.
- After the cells reach 50% confluence, switch to the fresh culture medium (complete mouse embryonic stem cell growth medium) containing 8 µg/mL polybrene).
- Add 100 µL of the lentiviral particle solution from step 10.3 including lenti-CRISPR V2-gRNAi22, lenti-iRFP670-gRNAi23 and control (empty vector: lenti-CRISPR V2, lenti-iRFP670) to mouse iPSCs. Incubate cells for 3 days at 37 °C with 5% CO2.
- Select stably infected cells with media containing 2.5 µg/mL blasticidin and 100 µg/mL hygromycin B by determining the minimum concentration of blasticidin and hygromycin B required to kill the un-infected cell.
NOTE: Un-infected cells would be killed by blasticidin and hygromycin B. - Digest the selected mouse iPSCs with 0.5 mL of TVP solution each well (24-well plate) and incubate cells for 30 min at 37 °C with 5% CO2.
- Dissociate the iPSCs into single cells by pipetting, count cells with a cell counting chamber and then dilute about 150 digested single cells with mES medium into 10 cm dish for culture at 37 °C with 5% CO2.
- After about 10 days, pick single colonies under an inverted microscope using 10 µL sterile pipette tips (96 colonies need to be picked).
- Transfer the picked colonies into 50 µL of TVP solution each well (96-well plate, one colony each well), digest at 37 °C for 30 min, and then seed the digested cells into two 96-well culture plates to keep culture (one for genotyping).
- Incubate in a CO2 incubator at 37 °C until 70% confluent.
12. Identification of iPSC colonies with exon23 deletion
- Remove the medium in 96-well plate when the cell colonies reach 70% confluence.
- Add 25 µL of lysis reagent (Table of Materials) containing proteinase K solution (1 mL of proteinase K in 100 mL of lysis reagent) to each well, and transfer the lysate to a 96-well PCR plate.
- Seal the PCR plate and incubate the plates at 55 °C for 30 min, and then at 95 °C for 45 min to lyse the cells and denature the proteinase K.
- Carry out PCR reaction with 2 µL of lysate from step 12.3. For the 20 µL PCR reaction, add 2 µL of lysate, 10 µL of 2x DNA polymerase premix (Table of Materials), 7 µL of DNase-free water, and 1 µL of DMD exon 23 primers (Table 1).
- Use the following parameters for PCR reaction: 98 °C for 1 min, 35 cycles of 98 °C for 10 s, 60 °C for 15 s, 72 °C for 30 s, and a final extension at 72 °C for 1 min.
- Load the PCR reaction onto a 2% agarose gel. Run the gel at 100−150 V for 30 min.
- Inspect the gel under UV light (the knockout efficiency is 3/94).
13. Using the Tet-on MyoD activation system to directly differentiate iPSC into myogenic progenitor cells (MPC)
- Package the LV-TRE-VP64-mouse MyoD-T2A-dsRedExpress2 and LV-TRE-VP16 mouse MyoD-T2A-dsRedExpress2 (a gift from Charles Gersbach) (Table of Materials) as previously described for lenti-CRISPRv2-blast and lenti-gRNA-iRFP670 vectors in sections 9 and 10.
- Infect mouse iPSC with lentivirus-TRE-VP64-MyoD-T2A-dsRed-Express2 or lentivirus-TRE-VP16-MyoD-T2A-dsRedExpress2 as previously described for lenti-CRISPRv2-blast and lenti-gRNA-iRFP670 vectors in steps 11.1−11.3.
- Select cells with 1 μg/mL puromycin after three days of infection to obtain a pure transduced cell population.
- Add 3 μg/mL doxycycline into culture media (10% FBS DMEM) to MPC differentiation. Replace fresh medium supplemented with doxycycline every two days.
14. Quantitative reverse transcription PCR for evaluating dynamic muscle differentiation and DMD exon 22-24 expression
- Extract cellular RNA after 0, 3, 6, and 10 days after doxycycline treatment using RNA isolation reagent, reversely transcribe RNA into cDNA by using first strand cDNA synthesis kit (Table of Materials).
- For the 20 µL qPCR reaction system, add 1 µL of cDNA, 10 µL of PCR reaction buffer (Table of Materials), 8 µL of DNA H2O, and 1 µL of mixture of forward and reverse primers (glyceraldehyde-3-phosphate dehydrogenase [GAPDH], skeletal muscle [ACTA1], OCT4 and DMD exon22, DMD exon23, and DMD exon24, see Table 1).
- Use the following parameters for PCR reaction: 50 °C for 2 min, 95 °C for 2 min, 40 cycles of 95 °C for 15 s, 60 °C for 1 min, melt curve 65.0 °C to 95.0 °C, increment 0.5 °C.
15. Immunofluorescence staining of myosin heavy chain 2 (MYH2) and dystrophin protein expression
- Plate doxycycline-induced, lenti-TRE-MyoD modified cells from step 13.4 onto 8-well culture slides.
- Fix cells in 4% formaldehyde for 15 min at room temperature, and then wash the slides twice in PBS for 5 min each time.
- Block the cells with 5% goat serum protein diluent for 1 h at room temperature.
- Add rabbit-anti-dystrophin antibody (1:300) and mouse-anti-MYH2 antibody (1:100) to the slides, incubate at 4 °C overnight in a humidified chamber.
- Discard the primary antibody solution, wash the cells 3x (5 min/wash) in PBS, add 1:400 diluted Alexa488-conjugated goat-anti-rabbit antibody and Alexa555-conjugated goat-anti-mouse antibody to slides, and incubate for 45 min at room temperature.
- Wash the slides 3x (5 min/wash) in PBS, and mount sections with mounting medium containing DAPI.
Establishment of Dmdmdx skin fibroblasts derived iPSC. We demonstrated the efficiency of generating mouse iPSCs from Dmdmdx mice derived skin fibroblast using the integration-free reprogramming vectors. Figure 1A demonstrated that the appearance of embryonic stem cell (ESC)-like colonies at three weeks after infection. We evaluate the efficiency of iPSC induction by live alkaline phosphatase (AP) stain; Figure 1B shows that the percentage of AP-positive cells was around 1.8% by FACS analysis. SSEA1, Lin28, Nanog, OCT4 and SOX2, pluripotency markers for mouse embryonic stem cells, were positive for iPSC colonies by immunofluorescent staining, (Figure 1C). To investigate the three germline differentiation of iPSCs in vivo, we intramuscularly injected iPSCs into the mouse gastrocnemii. We observed that the injected iPSCs differentiated into liver cells (endoderm), smooth muscle cells (mesoderm), and adrenergic neuron cells (ectoderm) (Figure 1D), indicting the pluripotency of iPSCs.
CRISPR/Cas9-mediated exon23 deletion. We designed two guide RNAs that flank the mutant exon 23. After Cas9-mediated double-stranded breaks (DSB) and non-homologous end joining (NHEJ), mutant exon 23 was deleted, allowing for truncated but functional dystrophin production (Figure 2A). To identify exon 23 deleted mouse iPSC, cells were sparsely seeded, and individual colonies were picked and propagated. Genomic DNAs extracted from these colonies were subjected to PCR genotyping. Figure 2B demonstrated that colony #1 and #2 have exon 23 deletions indicating a successful deletion of the exon 23.
Differentiating mouse iPSCs into a myogenic lineage and restoring dystrophin expression. We use a tetracycline-inducible MyoD expression system to induce myogenic differentiation of iPSCs. Doxycycline was used to induce MyoD expression in iPSCs. Figure 3A shows the time course of muscle differentiation in Dox-treated iPSCs. qRT-PCR showed that the mRNA level of OCT4, a pluripotent marker, gradually decreased, while the expression of ACTA1, a skeletal muscle marker, increased after Dox induction. Also, we observed the myotubes formation at two weeks after Dox treatment (Figure 3B). Importantly, the qRT-PCR assay showed the recovery of DMD exon 24 mRNA expression in Dox-induced, Cas9-mediated Exon23 deleted line in comparison to Cas9-control line (Figure 3C). Inconsistent with qRT-PCR, immunofluorescent staining shows the dystrophin protein expressionin Cas9-mediated exon 23 deleted cells, whereas the dystrophin expression was absent in control cells (Figure 3D).
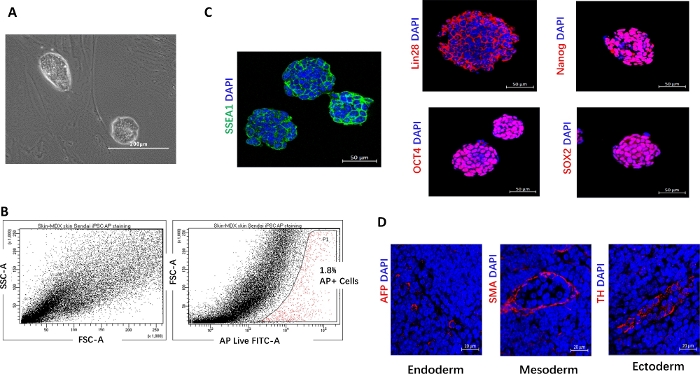
Figure 1: Reprogramming skin fibroblasts from Dmdmdx mice into iPSCs.
(A) Representative image of ES-like colonies (scale bar = 200 µm). (B) FACS analysis of the reprogramming efficiency of mouse skin fibroblasts into iPSCs after 8 days of Sendai virus transduction by live AP staining. (C) Immunofluorescent staining of SSEA1, Lin28, Nanog, Oct4, and SOX2 in iPSCs (scale bar = 50 µm). (D) Immunofluorescent staining for AFP (endoderm), SMA (mesoderm), and tyrosine hydrolase (TH) (ectoderm) of teratoma 2 weeks after iPSC injection into gastrocnemii (scale bar = 20 µm). Please click here to view a larger version of this figure.
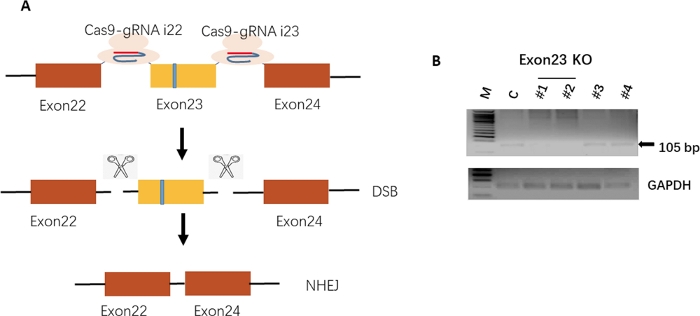
Figure 2: CRISPR/Cas9-mediated exon23 deletion.
(A) Schematic diagram of CRISPR/Cas9-mediated exon 23 deletions. The Cas9 nuclease targets intron 22 and intron 23 by two gRNAs. Double-stranded breaks (DSBs) by Cas9 results in the excision of the mutant exon 23. The distal ends are repaired by non-homologous end joining (NHEJ), resulting in the restoration of the reading frame of the dystrophin gene. (B) PCR genotyping analysis of exon 23. The arrow indicates the PCR product of exon 23. GAPDH serves as a reference. Please click here to view a larger version of this figure.
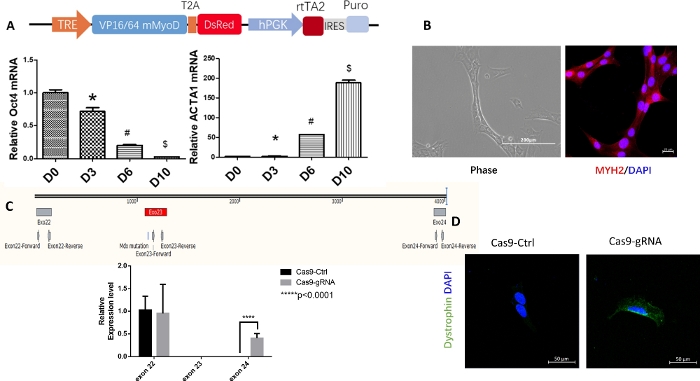
Figure 3: Differentiating mouse iPSCs into the myogenic lineage and restoring dystrophin expression.
(A) qRT-PCR showed the time course of mRNA level of Oct4 and ACTA1 in Dox-treated exon 23-deleted Dmdmdx iPSC (*P < 0.05 vs D0, D6, D10, #P < 0.05 vs D0, D3, D10, $P < 0.05 vs D0, D3, D6, n = 4 for Oct4) (*P < 0.05 vs D6 and D10, #P < 0.05 vs D0, D3, and D10, $P < 0.05 vs D0, D3, and D6, n = 3 for ACTA1). (B) Left: Representative image of myotube formation from Dox-induced mouse iPSCs (scale bar = 200 µm). Right: Immunofluorescent analysis of MYH2 in myotube formation from Dox-induced mouse iPSCs (scale bar = 20 µm). (C) Upper: the PCR primer positions for DMD Exon22, Exon23 and Exon24; Bottom: qRT-PCR analysis of the mRNA level of DMD Exon22, Exon23, and Exon24 expression in MPC (****P < 0.0001, n = 3). (D) Immunofluorescent analysis of dystrophin expression in Dox-induced MPC from iPSCCas9-Ctrl and iPSCCas9-gRNA (scale bar = 50 µm). Please click here to view a larger version of this figure.
| Guide primers | |
| i22 sense | 5'-CACCGTTAAGCTTAGGTAAAATCAA- 3' |
| i22 antisense | 5’-AAACTTGATTTTACCTAAGCTTAAC-3’ |
| i23 sense | 5'-CACCGAGTAATGTGTCATACCTTCT- 3' |
| I23 antisense | 5’-AAACAGAAGGTATGACACATTACTC-3’ |
| PCR primers | |
| OCT4-Forward | 5'-AGCTGCTGAAGCAGAAGAGGATCA-3' |
| OCT4-Reverse | 5'-TCTCATTGTTGTCGGCTTCCTCCA-3' |
| ACTA1-Forward | 5'-GATCCATGAGACCACCTACAAC-3' |
| ACTA1-Reverse | 5'-TCAGCGATACCAGGGTACAT-3' |
| Exon22-Forward | 5'-TTACCACCAATGCGCTATCA-3' |
| Exon22-Reverse | 5'-CCGAGTCTCTCCTCCATTATTTC-3' |
| Exon23-Forward | 5'-CCAAGAAAGCACCTTCAGAAATATG-3' |
| Exon23-Reverse | 5'-TTTGGCAGCTTTCCACCA-3' |
| Exon24-Forward | 5'-AAC CTT ACA GAA ATG GAT GGC-3' |
| Exon24-Reverse | 5'-TTTCAGGATTTCAGCATCCC-3' |
| GAPDH-Forward | 5'-TGACAAGCTTCCCATTCTCG-3' |
| GAPDH-Reverse | 5'-CCCTTCATTGACCTCAACTACAT-3' |
Table 1: Primer sequence.
