It is important to thaw frozen Drosophila cells rapidly and culture them at a cell density that brings the culture back into the growth phase. If the procedures for cryopreservation and thawing are adhered to, the cell density in the T-25 flask will at least equal 4 x 106 cells/mL. One to two hours after thawing, most Drosophila cell lines will begin to attach to the growing surface. Under the circumstance in which most of the cells have not attached on the growing surface within two hours after thawing, it is recommended to incubate the cells overnight before changing the media.
The goal of subculturing is to maintain cells in the healthy exponential log-phase of the growth curve. The criteria for subculturing depend on the visible lack of microorganismal contamination, cell density, and the need to establish a regular maintenance schedule. It is important to first assess the health of the cells and determine the absence of adventitious contaminants prior to freezing. Most bacterial and fungal contaminants are easy to detect simply by visual inspections. Contaminated cultures can be identified by an increase in media turbidity. Under the microscope, contaminants may appear as bacterial rods, cocci, budding yeast cells or string-like fungal hyphae. Other sources of contamination such as the non-cytopathic mycoplasma cannot be visually detected and can be routinely tested by PCR-based assays21.
The confluence of a cell line can be determined visually (Figure 1). Fast growing cell lines reach confluence early and need to be passaged regularly. Such lines are subcultured up to twice a week. In contrast, slow growing cells are passaged at least once every two weeks or longer. However, the cells need to be fed fresh media every week. This is to prevent media exhaustion and to dilute metabolic waste products from the cells. Cell lines derived from varying tissue sources differ in their morphology (Figure 2), adherence properties, media requirements (Table 1) and doubling time (Table 2). Table 5, Table 6, Table 7, and Table 8 list the recipes for the various Drosophila cell culture media.
Cell counting ensures an accurate seeding density and a predictable routine for subculturing. For quantitative experiments, cell counting is essential. Cells are counted either by using a hemocytometer (Figure 3A) or an automated particle counter (Figure 3B). If using an automated counter, follow the manufacturer's instructions. Counting cells manually using a hemocytometer is economical and easy. The number of cells enclosed in the middle Neubauer grids are counted and the cell density is calculated; For example, n = 214 cells, resulting in a cell density of 2.14 x 106 cells/mL (Figure 3D).
Cell suspension from two 100 mm plates, each containing 10 mL of cell suspension at 4 x 106 cells/mL are collected and resuspended in 2 mL of freezing media to achieve a density of 4 x 107 cells/mL. Each frozen cryovial with 0.5 mL of cell suspension contains 2 x 107 cells. This will result in a culture with 4 x 106 cells/mL when thawed according to the protocol section 1.
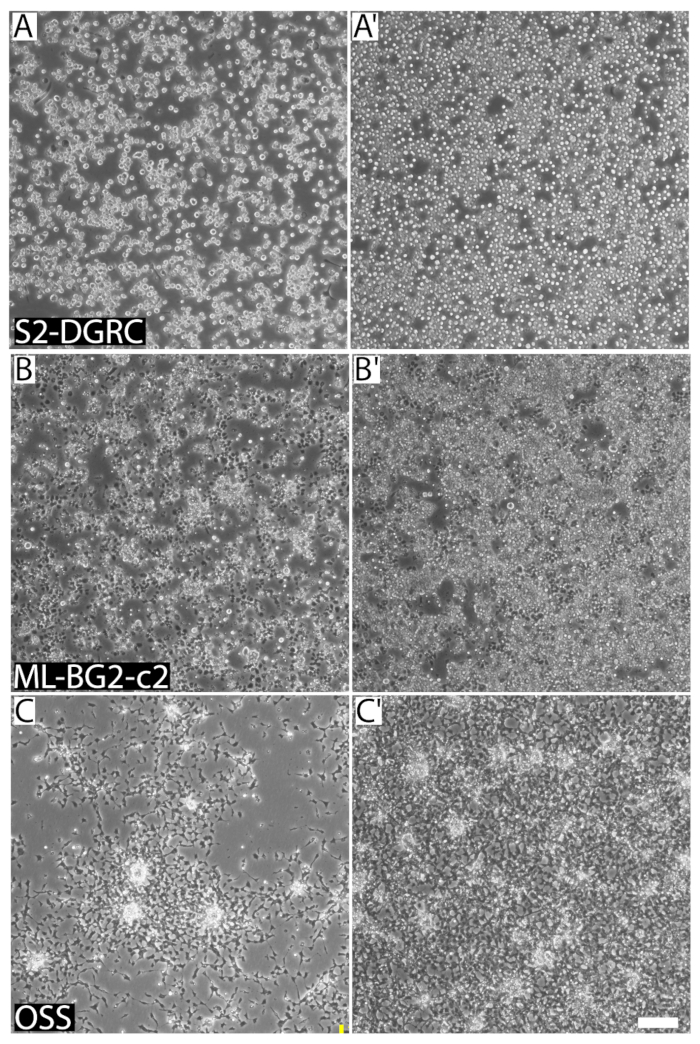
Figure 1: Representative images of three distinct Drosophila cell lines at different confluence and cell densities. (A) S2-DGRC culture at 1 x 106 cells/mL. (A') S2-DGRC culture at 4.5 x 106 cells/mL. (B) ML-BG2-c2 culture at 2 x 106 cells/mL. (B') ML-BG2-c2 culture at 8 x 106 cells/mL in which cells are piling and aggregating as foci. (C) OSS culture at 1 x 106 cells/mL. (C') OSS culture at 4 x 106 cells/mL. Cells in suspension are not captured on the same focal plane. Scale bar = 100 µm. Please click here to view a larger version of this figure.
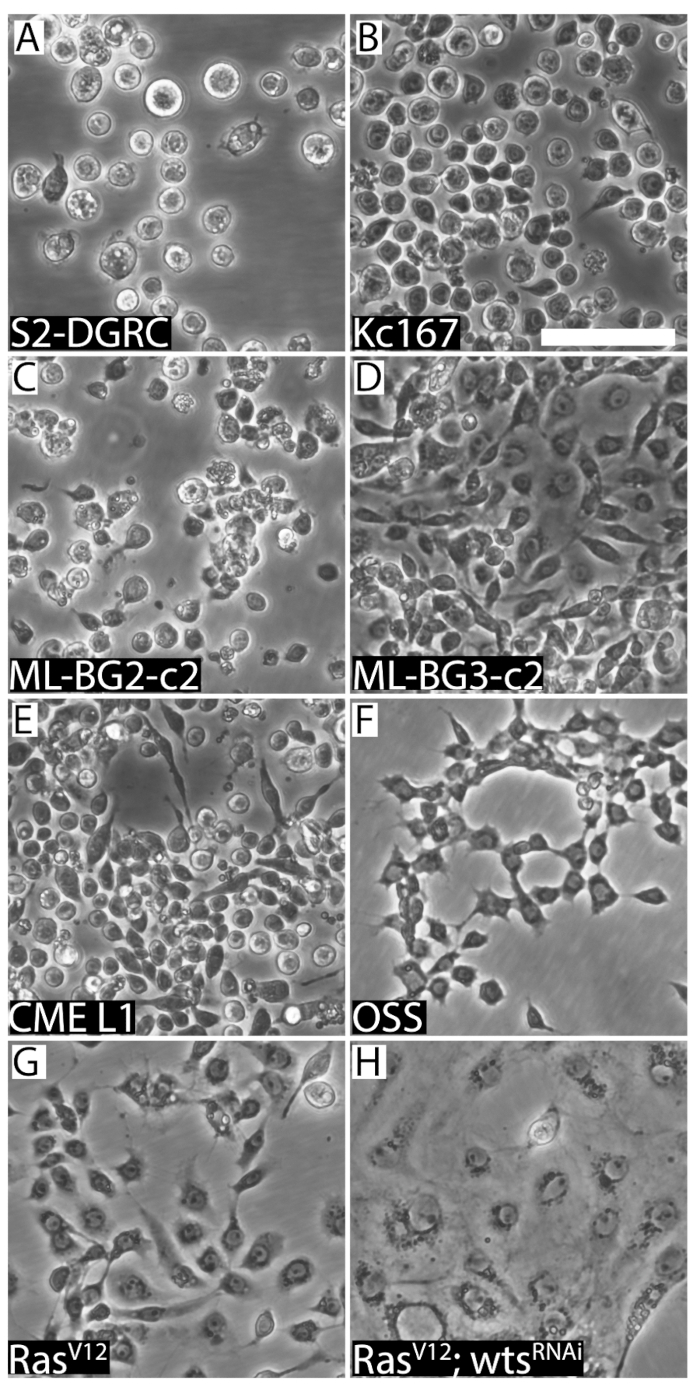
Figure 2: Representative images of the eight distinct Drosophila cell lines. (A) Round embryo-derived S2-DGRC. (B) Round embryo-derived Kc167. (C) Round larval CNS-derived ML-BG2-c2. (D) Round larval spindle-shaped ML-BG3-c2. (E) CME L1, a cell line derived from the larval leg imaginal discs, is smaller and has round/fusiform morphology. (F) OSS, a cell line derived from adult ovaries, displays spindle-shaped morphology. (G) Spindle-shaped RasV12 cell line expressing activated Ras. (H) RasV12; wtsRNAi (WRR1), a cell line expressing activated Ras and double-stranded RNA targeting the tumor suppressor warts (wts), displays epithelial characteristics. Scale bar = 50 µm. Please click here to view a larger version of this figure.
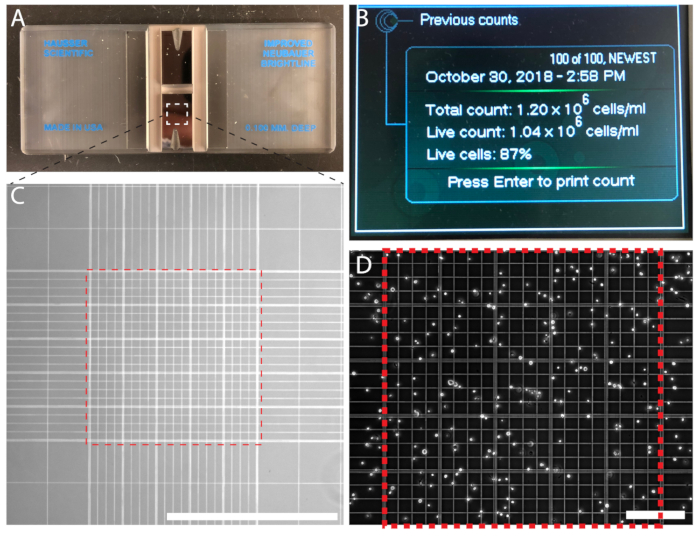
Figure 3: Cell density can be counted manually using a hemocytometer or automatically using an automated particle counter. (A) A hemocytometer with two chambers. (B) An automated cell counter displaying the output of a cell count. (C) The improved Neubauer cell counting grid viewed under a 10x objective. Count cells bound by the 0.1 mm3 central grid (red dashed-line square). (D) The central grid on the hemocytometer filled with cells for counting. Scale bar = 1 mm (C); 0.2 mm (D). Please click here to view a larger version of this figure.

Figure 4: Equipment for cryopreservation. (A) A freezing container stores the ampules in an upright position for slow freezing. (B) A metal cane for holding frozen ampules. (C) A canister for holding canes. (D) A plastic freezing box (cryobox). (E) A canister holding multiple canes inserted into a liquid N2 storage tank. (F) A liquid N2 storage tank. Please click here to view a larger version of this figure.
| Cell Strain | Media | Adherence | Trypsin |
| Schneider Lines | M3 + BPYE + 10% fetal calf serum (FCS), pH 6.6 | Semi-adherent | No |
| (S2R+, S2-DRSC, S2-DGRC, Sg4)11 | |||
| Schneider’s media+ + 10% FCS | |||
| Kc lines (Kc167, Kc7E10)21,22 | M3 + BPYE + 5% FCS, pH 6.6 | Semi-adherent | No |
| Hyclone-CCM3, pH 6.2 | |||
| Imaginal disc and CNS lines (ML-lines)13,14 | M3 + BPYE + 10% FCS, pH 6.6 | Semi-adherent | No |
| 10 µg/mL insulin | |||
| Milner imaginal disc lines (CME-lines)15 | M3 + 2% FCS | Semi-adherent | No |
| 5 µg/mL insulin | |||
| 2.5% fly extract | |||
| fGS/OSS16 | M3 + 10% FCS, pH 6.8 | Adherent | * |
| 10 µg/mL insulin | |||
| 1 mg/mL C5H8KNO4 | |||
| 0.5 mg/mL KHCO3 | |||
| 0.6 mg/mL glutathione | |||
| 10% fly extract | |||
| RasV12 lines18 | M3 + BPYE + 10% FCS, pH 6.6 | Adherent | Yes |
Table 1: The properties and media requirements of various Drosophila cell lines. Different isolates of semi-adherent Schneider lines including S2R+, S2-Drosophila RNAi Screening Center (DRSC), S2-Drosophila Genomics Resource Center (DGRC), and Sg4 are commonly used cell lines that proliferate robustly when cultured in M3 media + Bactopetone yeast extract (BPYE) supplemented with 10% fetal calf serum (FCS). Alternatively, Schneider's media (pH 6.7-6.8) is often used in place of M3+BPYE. The Kc lines proliferate in either M3 + BPYE (5% FCS) or serum-free CCM3 media. The ML imaginal disc and central nervous system (CNS) lines require insulin supplementation for proliferation. The Milner imaginal disc lines require both insulin and fly extract supplementation. Adherent fGS/OSS cell lines require insulin, a higher concentration of fly extract as well as glutathione for growth. Adherent RasV12 lines grow well in M3 + BPYE (10% FCS). Trypsin is used to dislodge adherent cell lines from the growth surface.
| Cell line (Stock #) | Genotype | Doubling time (h)* | Tissue Source |
| S2R+ (150) | OreR | 39 | Late embryos |
| S2-DGRC (6) | OreR | 23 | Late embryos |
| S2-DRSC (181) | OreR | 46 | Late embryos |
| Kc167 (1) | e/se | 22 | 6−12 h embryos |
| ML-BG2-c2 (53) | y v f mal | 48 | 3rd instar larval CNS |
| ML-BG3-c2 (68) | y v f mal | 104 | 3rd instar larval CNS |
| ML-DmD8 (92) | y v f mal | 66 | 3rd instar larval wing disc |
| CME W1 Cl.8+ (151) | OreR | 46 | 3rd instar larval wing disc |
| CME L1 (156) | OreR | 47 | 3rd instar larval leg disc |
| OSS (190) | bamD86 | 45 | Adult bam mutant ovaries |
| RasV12 lines | UAS-GFP; P(UAS-Ras85D.V12)/ P(Act5C-GAL4)17bFO1 | 41−65 | Embryo |
Table 2: Genotype, doubling time, and tissue sources of widely used Drosophila cell lines. The tissue genotype, source and population doubling time of commonly used cell lines are presented. Doubling time is based on growth in the recommended media at 25 °C.
| Culture vessel | Volume of media (mL) |
| 12.5 cm2 T-flask | 2.5 |
| 25 cm2 T-flask | 5 |
| 75 cm2 T-flask | 15 |
| 35 mm plate | 1 |
| 60 mm plate | 4 |
| 100 mm plate | 10 |
| 384-well plate* | 0.04/well |
| 96-well plate* | 0.1/well |
| 48-well plate* | 0.3/well |
| 24-well plate* | 0.5/well |
| 12-well plate | 1.0/well |
| 6-well plate | 2.0/well |
Table 3: Culture vessels and the recommended media volumes. Culture vessels of various sizes are available for culturing Drosophila cells. The appropriate media volumes (mL) are recommended for each vessel. Seal multi-well plates containing less than 0.5 mL of cell suspension with paraffin film to reduce media loss due to evaporation.
| Volume | |
| M3 + BPYE, pH 6.6 | 70 mL |
| Heat inactivated FCS | 20 mL |
| Sterile filtered DMSO* | 10 mL |
Table 4: Recipe for preparing 100 mL of freezing medium (M3 + BPYE, 20% FCS, 10% DMSO). Prepare freezing media as needed and avoid storing freezing media containing DMSO for prolonged period.
| M3 + BPYE medium | Amount |
| Shields and Sang’s M323 | 1 bottle |
| KHCO3 | 0.5 g |
| Select yeast extract | 1.0 g |
| Bactopeptone | 2.5 g |
| Sterile purified water | 1000 mL |
Table 5: Recipe for preparing 1 L of M3 + BPYE tissue culture medium. Adjust pH to 6.6. Sterilize by passing the medium through a 0.22 µm filter.
| Base M3 medium for fGS/OSS cell line | Amount |
| Shields and Sang M3 | 1 bottle |
| KHCO3 | 0.5 g |
| C5H8KNO4 | 1.0 g |
| Sterile purified water | 1,000 mL |
Table 6: Recipe for 1 L of fGS/OSS M3 base medium. Adjust pH to 6.8. Sterilize by passing the medium through a 0.22 µm filter.
| Hyclone-CCM3 | Amount |
| CCM3 powder | 28.6 g |
| NaHCO3 | 0.35 g |
| 10 N NaOH | 2.5 mL |
| CaCl2 | 0.5 g |
| Sterile purified water | 1,000 mL |
Table 7: Recipe for 1 L of Hyclone-CCM3 tissue culture medium. Adjust pH to 6.2. Sterilize by passing the medium through a 0.22 µm filter.
| M3 + BPYE + 10% FCS | Miyake disc and CNS lines medium | Milner disc lines medium | fGS/OSS complete medium | |
| M3 + BPYE, pH 6.6 | 90 mL | 90 mL | – | – |
| Heat inactivated FCS* | 10 mL | 10 mL | 2 mL | 10 mL |
| Insulin (10 mg/mL) | – | 100 µL | 50 µL | 100 µL |
| Fly extract | – | – | 2.5 mL | 10 mL |
| Glutathione (60 mg/mL) | – | – | 1 mL | |
| M3, pH 6.6 | – | – | 97.5 mL | – |
| fGS/OSS M3, pH 6.8 | – | – | 79 mL |
Table 8: Recipe for preparing 100 mL of various common Drosophila cell culture media. Incubate FCS at 56 °C for 1 h and shake every five minutes to heat-inactivate complement proteins.
