Commercially available oxygen scavenger PCD is frequently contaminated with a DNA nuclease. Contaminating nuclease activity could lead to spurious results in fluorescent studies, particularly studies that analyze DNA or DNA interacting proteins.We have found that recombinant PCD, a heterodimer of hexahistidine tagged pcaH and pcaG, may be expressed in E. coli (Figure 1). The heterodimer is first purified by nickel affinity chromatography (Figure 2). PCD is eluted in 2 steps of imidazole concentrations. Chromatography fractions are analyzed by SDS-PAGE. Fractions of nearly pure PCD are concentrated and further purified by SEC (Figure 3). SEC fractions are individually analyzed for both PCA oxidation activity and nuclease activity (Figure 4). Fractions that displayed high oxidation activity and no apparent nuclease activity are assayed for protein concentration and kept in a -80 °C freezer for experimental use.
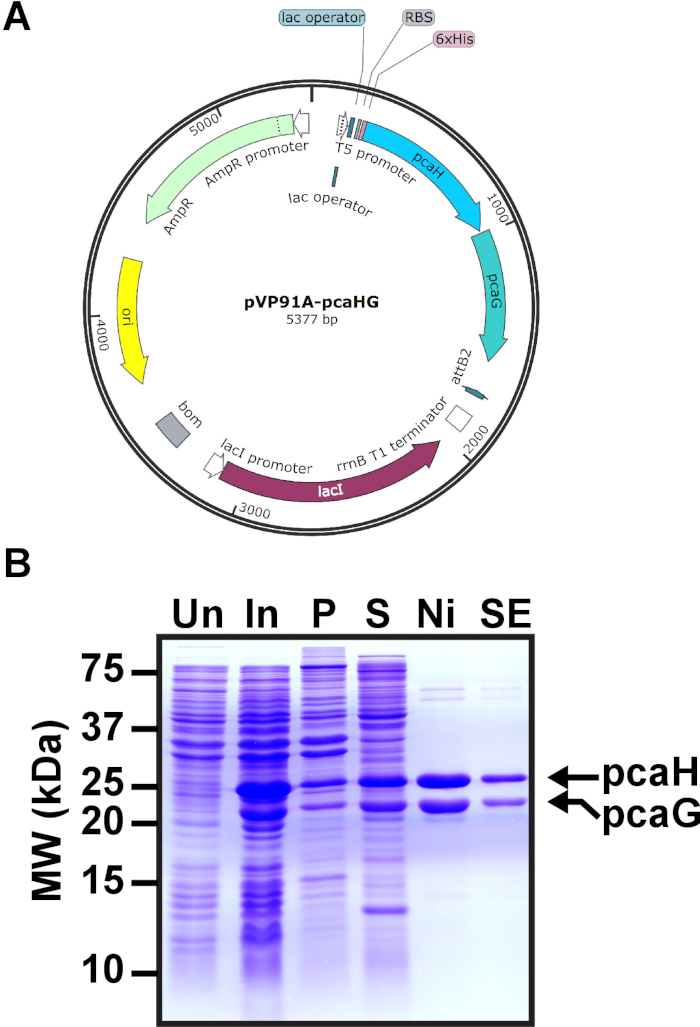
Figure 1: Induction of PCD in E. coli. (A) pVP91A-pcaHG is shown with the pcaG (α) and hexahistidine-tagged pcaH (β) PCD subunits. (B) Representative SDS-PAGE gel of PCD induction. Molecular weights are indicated on the left. The mobilities of 28.3 kDa hexahistidine-tagged pcaH and 22.4 kDa pcaG are on the right. Uninduced E. coli (Un), induced E. coli (In), the pellet following E. coli lysis and ultracentrifugation (P), the supernatant following ultracentrifugation to be loaded to a nickel column (S), representative fraction following nickel chromatography (Ni), and representative fraction following SEC (SE). This figure has been modified from a previous publication12. Please click here to view a larger version of this figure.
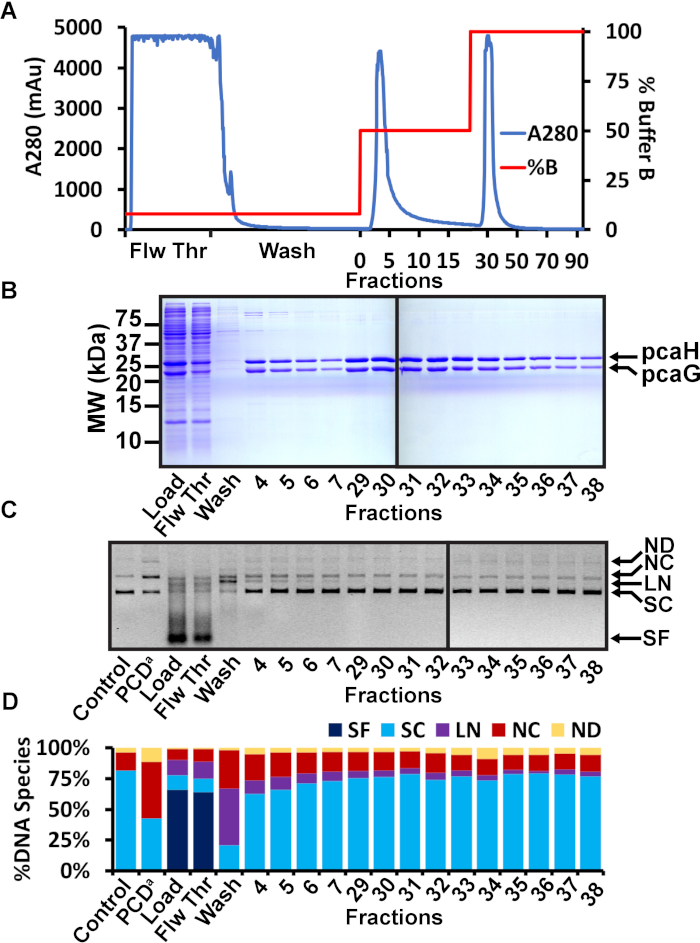
Figure 2: Nickel affinity chromatography purification of PCD. (A) Chromatogram of nickel affinity chromatography of PCD. The A280 is shown in blue and the percent concentration of Ni Buffer B is shown in red. The sample was loaded in a low 20 mM imidazole concentration. The flowthrough (Flw Thr) shows the soluble bacterial proteins that did not bind to the nickel resin. The column was washed with 20 mL of 20 mM imidazole buffer. A second 15 mL wash was performed with 125 mM imidazole. Elution of PCD was performed with 250 mM imidazole. Some PCD eluted in the presence of 125 mM imidazole, but the majority of the protein eluted in 250 mM imidazole. (B) Representative SDS-PAGE analysis of nickel affinity fractions. The load, flowthrough (Flw Thr), and first wash showed the successful induction of PCD, the soluble bacterial proteins that did not bind the nickel resin, and the minimal proteins observed during the first wash, respectively. Several fractions throughout the second wash and elution steps are shown. Fractions from the second wash included PCD protein but also displayed detectable higher molecular weight contaminants. Fractions from the elution step appeared to be free of contaminants. Molecular weights are shown on the left. Mobilities of pcaH and pcaG are shown on the right. (C) Agarose gel of nuclease assay. The nickel affinity column load, flowthrough, wash, and multiple fractions were tested for nuclease activity. A negative control (control) is the plasmid without added protein. A positive control (PCDa) is a commercially available PCD known to be contaminated with a DNA nuclease. DNA species are indicated on the right as small fragments (SF), supercoiled (SC), linear (LN), nicked circle (NC), and nicked dimer (ND). (D) Quantitation of the various DNA species observed in the agarose gel nuclease assay. The total pixel volume of each lane was measured. The pixel volume of each DNA species was determined and expressed as a percentage of the total pixel volume in the lane. The negative control was 81.7% supercoiled with 14.4% nicked circles. The positive control displayed a significant increase of 46.0% nicked circles. The load and flowthrough contained bacterial nucleases that converted the plasmid and contaminating bacteria DNA to small fragments. The first wash at 20 mM imidazole also appeared to contain significant nuclease activity, resulting in linear and nicked circles. Fractions 4-7 from the second wash at 125 mM imidazole also displayed significant nuclease activity (particularly, fractions 4 and 5 that generated observed linearized plasmid). Fractions 29-38 from the elution step appeared more similar to the negative control. In this example, fractions 29-38 were chosen to be combined, concentrated, and further purified by SEC. This figure has been modified from a previous publication12. Please click here to view a larger version of this figure.
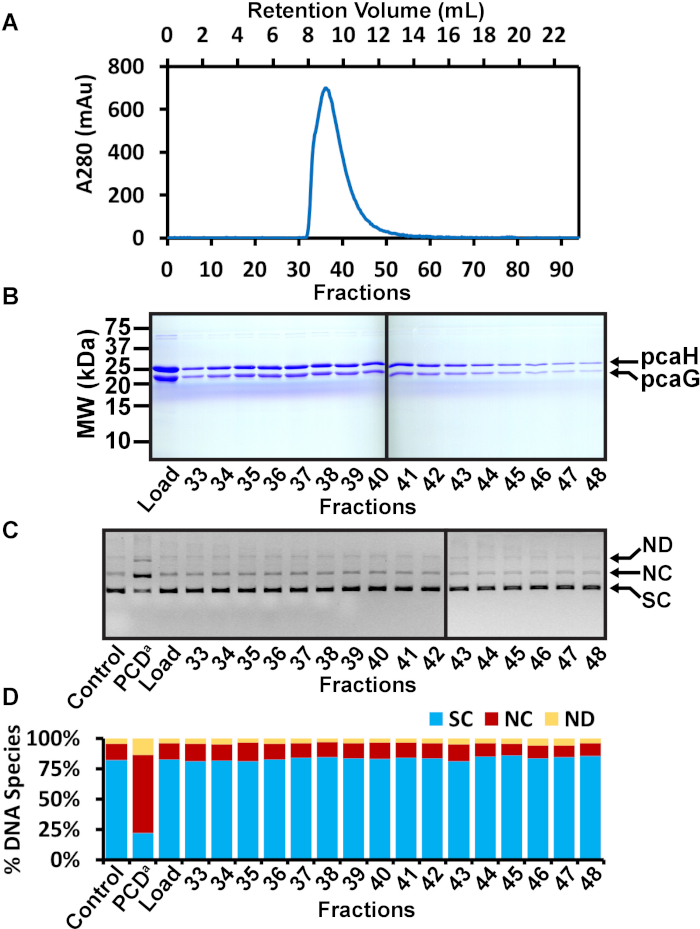
Figure 3: SEC purification of PCD. (A) Chromatogram of SEC of PCD fractions following nickel affinity chromatography. The A280 is shown in blue and elution fractions are indicated. PCD eluted from SEC as a single apparent peak. (B) Representative SDS-PAGE analysis of SEC fractions 33-48. The load is the concentrated PCD following nickel affinity purification. Fractions 33-48 span the apparent SEC peak. No detectable contaminants were observed. (C) Agarose gel of nuclease assay. The SEC load and multiple fractions were tested for nuclease activity. A negative control (control) is the plasmid without added protein. A positive control (PCDa) is a commercially available PCD known to be contaminated with a DNA nuclease. DNA species are indicated on the left as supercoiled (SC), nicked circle (NC), and nicked dimer (ND). (D) Quantitation of the various DNA species observed in the agarose gel nuclease assay. The total pixel volume of each lane was measured. The pixel volume of each DNA species was determined and expressed as a percentage of the total pixel volume in the lane. The negative control was 82.1% supercoiled with only 13.7% nicked circles. The positive control displayed a significant increase of 64.8% nicked circles. The SEC load displayed no apparent nuclease activity due to judicious choice of fractions from the nickel affinity purification. Similarly, fractions 33-48 appeared similar to the negative control. For example, fraction 36 was 82.5% supercoiled and 13.2% nicked circle. In this example, fractions 36 and 37 were chosen to be quantified, frozen, and kept in a -80 °C freezer for future experimental use. This figure has been modified from a previous publication12. Please click here to view a larger version of this figure.
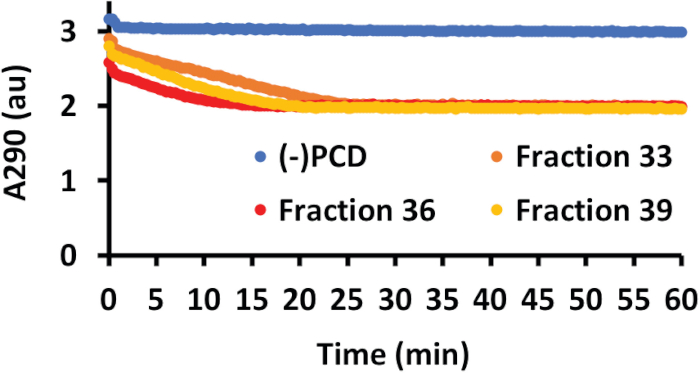
Figure 4: PCA oxidation and nuclease activity of PCD SEC fractions. PCA oxidation was measured by A290. As PCD oxidized the PCA molecule, the A290 decreased. PCA oxidation was measured every 20 s for 1 h. A negative control with no added PCD fraction (blue line) showed no change in A290, indicating the PCA molecule was stable. Data from three representative SEC fractions (36 in red, 33 in orange, 39 in yellow) show that purified PCD reduced the A290, indicating oxidation of PCA. This figure has been modified from a previous publication12. Please click here to view a larger version of this figure.
