To demonstrate the effectiveness of the multilayer microfluidic platform for the conduction of IVTT experiments, the described setup was used to express the deGFP protein. The experiment was conducted in a commercially available30 IVTT reaction mixture – comprising all the necessary transcription and translation componentry – supplemented with reaction substrates and DNA templates. Experiments were conducted at a temperature of 29 °C; a temperature found to be optimal for the IVTT expression of proteins.
The microfluidic device possesses nine unique inlets, of which four were utilized during this experiment. The first contained the commercially obtained IVTT reaction mixture. The IVTT reaction mixture accommodates all the components required to successfully express proteins however, purified GamS was added to the reaction mixture – at a final concentration of 1.3 µM – prior to loading into the microfluidic device. The addition of the GamS protein serves to minimize the degradation of linear DNA species when performing the experiments. Crucially, the IVTT mixture was injected into polytetrafluoroethylene (PTFE) tubing coiled onto a Peltier element with a surface temperature of 4 °C to cool the solution prior to the injection thereof into the microfluidic device; preventing the degradation of the reaction solution prior to its use. Micro-bore polyether ether ketone (PEEK) tubing was used to connect the PTFE tubing leaving the Peltier element surface with the microfluidic device, reducing the volume of the IVTT reaction mixture not being cooled. The second solution inserted into the device contained the linear DNA template coding for the deGFP – dissolved in ultrapure water – at a concentration of 10 nM. The third solution, ultrapure water, served multiple purposes during the experimental procedures. Primarily, the ultrapure water was used to ensure that the displaced volume per dilution was equal for all reactors, acting as a replacement for DNA in the control reactions. Additionally, ultrapure water was also used to dilute the fluorophore during the device calibration and to flush the dead volume of the device when switching between reagents. The final solution inserted into the device was a purified FITC-dextran solution (25 μM) required to perform the initial device calibration. The DNA, water, and fluorophore solutions were injected into tubing (0.02” ID, 0.06” OD) which could subsequently be inserted into one of the inflow channels of the microfluidic device as per Section 4.2 of the protocols. As such, these solutions were stored at 29 °C for the entirety of the experiments.
The actuation of the control channels of the microfluidic device is achieved via custom control software where each of the control channels can be individually actuated. The execution of prolonged IVTT reactions cannot be achieved via this manual process and requires the use of automated protocols incorporated within the control software. When preparing a microfluidic device for experiments, similar automated protocols can be utilized to execute a number of useful processes: the flushing of the device dead volume with a new reagent, the mixing of the reagents within the ring reactor, and the loading of a new reagent into the reactor whilst displacing an equal volume of the current solution. In addition, two complex process are available: the conduction of a device calibration, and the execution of a prolonged cell-free protein expression. All of the aforementioned processes can be easily executed from the main interface, alongside the ability to configure multiple parameters to vary specific process settings such as the inflow channel, inflow volume, and mixing duration.
Due to fluctuations in pressure and imperfections during microfluidic device fabrication, the volume of fluid displaced during a single injection cycle can vary between devices. As such, prior to performing IVTT experiments, the displaced reactor volume per injection cycle (Refresh Fraction) was determined. This calibration requires the filling of all eight reactors with a fluorescent reference solution. In this case, a purified FITC-dextran solution (25 μM) was used. Subsequently, the reactors are diluted 10 times with ultrapure water. By measuring the decrease in fluorescence per dilution cycle for each reactor, the volume of fluid displaced during a single injection cycle was determined. Within the control software, this value (the Refresh Ratio) was recorded for use during the IVTT experiment. Crucially, to account for variations in the flow rate across the device, as well as discrepancies in the individual reactor volumes, the Refresh Ratio is determined and stored for each individual reactor. The sequence of filling and diluting the reactors was conducted automatically using the Perform Calibration program which forms part of the control software. The results of the calibration experiment are shown in Figure 8.
The most complex pre-programmed process executes a long-duration IVTT experiment, allowing users to initiate the experiment and subsequently allow it to operate unattended until completion. Throughout the experiment, reactors 1 and 5 were used as blanks, with only water being added to the reactors during dilutions. Reactors 2 and 6 were utilized as negative controls and contained only IVTT reaction solution and ultrapure water. The remaining reactors (3, 4, 7, and 8) contained the IVTT reaction solutions and 2.5 nM of linear DNA coding for the deGFP gene. Initialisation of the reactors is achieved by fully filling all the reactors (excluding 1 and 5) with the IVTT reaction solution, before 25% of the reactor volume was displaced with ultrapure water. Hereafter, the periodic injection of reagents into the reactors was initiated. The experiment was conducted such that new reagents were injected into the reactors every 14.7 minutes, with 30% of the reactor volume being displaced during each dilution cycle. The composition of each injection was such that 75% of the injected fluid comprised fresh IVTT solution, whilst the remaining 25% consisted of either DNA or ultrapure water. Following each injection of new reagents the reactors were continuously mixed, after which a fluorescence image of each reactor was recorded using the microscope. The reaction was subsequently allowed to run continuously for 68 cycles, resulting in an experimental duration of 16.5 h. The results of this experiment are given in Figure 9.
When performing prolonged IVTT experiments, there are two main causes for the failure of a reaction; the introduction of air into the microfluidic device or the degradation of the IVTT reaction solution. The occurrence of air within the microfluidic device is most often the direct result of small air bubbles existing in the inflow solutions, which are subsequently injected into the microfluidic device. Upon entering the device, the presence of air inhibits the proper flow of fluids, whereby the reactions are no longer periodically refreshed leading to the formation of batch reactions within the reactor rings. In some cases, the air is slowly removed from the device by the repeated flushing of reagents, after which the reaction continues as intended (as shown in Figure 9). In other cases the air remains trapped, and can only be removed by aborting the experiment and subsequently applying continuous (high) pressure to the flow layer of the microfluidic device, analogous to the filling process described in Section 5.1 of the protocols. During our experiments the cell lysate is stored in PTFE tubing on a Peltier element cooled to 4 °C. Both measures aid in limiting the degradation of the IVTT reaction solution over time, with the inert PTFE tubing ensuring limited interaction between the tubing and the reaction solution and the cold temperatures preserving the functional (bio)molecular componentry required to perform IVTT. Should degradation of the reaction solution occur – as the result of insufficient cooling or undesired interactions between the reaction solution and the storage environment – then this will exhibit itself experimentally as a gradual reduction of protein expression over time. Once degraded, the IVTT reaction solution cannot be recovered and a new experiment should be prepared.
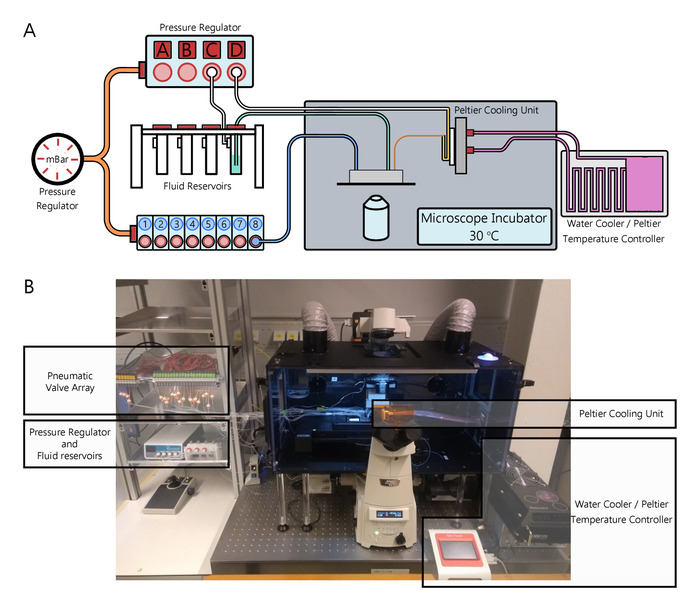
Figure 1. The hardware setup required to perform continuous IVTT reactions. A) Schematic of the hardware setup. B) Photograph of the setup used throughout this manuscript. The implementation of a multilayer microfluidic device for continuous IVTT reactions requires an extensive hardware setup to regulate flow pressure, actuate control channels, heat and cool reactions and reagents, store fluids, and image the device during experiments. Experiments are performed at temperatures of 30 °C, which is achieved by placing the microscope within an incubator set to this temperature. To prevent deterioration of the IVTT reaction solution, it is stored within PTFE tubing coiled over the cold face of a Peltier element. The temperature of the Peltier element is set to 4 °C, with a water cooler and water block being used to maintain this temperature. Reagents which do not require cooling, are stored in fluid reservoirs outside of the microscope incubator. Constant pressure is applied to these reservoirs by a computer controlled pressure regulator. In this manner, the fluids are forced through the outlet tubing of the reservoirs, which connect directly to the inflow channels of the microfluidic device. Each of the control channels of the microfluidic device is connected to a pneumatic valve. The entire valve array is under constant pressure. Opening the valve, allows for pressurisation of the fluid within the tubing connecting the pneumatic valve to the control channel of the microfluidic device, thus opening and closing the PDMS membranes found within the microfluidic device. The pneumatic valves are opened and closed via a user interface which commands a fieldbus controller (not shown) to open and close specific pneumatic valves. Figure adapted from Yelleswarapu et al.22. Please click here to view a larger version of this figure.

Figure 2. Overview of the pneumatic valve setup and control channel connection. An 8-valve array is shown with three control channel connections fitted to valves 1, 2, and 3. Compressed air can be supplied to the valve array via 1/4” tubing. For the actuations of control channels two pressures are used: 1 bar for the lower pressure control channels (1, 2, and 3) and 3 bar for the higher pressure control channels (9 through 30, not shown here). The tubing can be filled with ultrapure water and inserted into one of the control channel inlets using a stainless steel connector pin. Please click here to view a larger version of this figure.

Figure 3. Overview of the commercial flow pressure regulator and reservoir system. A commercially available pressure regulator is used to inject fluids into the flow layer of the multilayer microfluidic device. Connecting the pressure controller to a computer allows for modulation of the pressure used to perform the fluid injections. Reagents can be stored in a fluid reservoir, which is directly connected to the pressure regulator. The application of pressure to the reservoir forces the fluid out of the reservoir via the outlet tubing. This outlet tubing can be connected directly to one of the fluid inlets of the microfluidic device using a stainless steel connector pin. In the event that the reagent volume is unable to reach the fluid reservoir, the outlet tubing acts as a reservoir for the reagent. Please click here to view a larger version of this figure.

Figure 4. Overview of the cooling system used to cool reaction reagents. (Left) Isolated cooling setup and (Right) Cooling setup placed within the microscope and connected to the microfluidic device. A Peltier element is used to cool the IVTT reaction solution prior to injection into the microfluidic device. The reagent is stored within PTFE tubing coiled over the cold-face of the Peltier element. A length of PEEK tubing is used to transfer the cooled fluid to the microfluidic device, with the small internal diameter (0.005”) minimizing the reagent volume no longer being cooled. Alongside the coiled PTFE tubing, a thermistor is placed, allowing for real-time temperature monitoring on the surface of the Peltier element. The voltage applied to the Peltier is set such that the surface temperature of the Peltier remains between 0 °C and 4 °C. To remove excess heat produced by the Peltier element, the hot-face of the Peltier is placed against a water cooled block, with the addition of silicone free heat sink grease ensuring optimal heat transfer between the two faces. Please click here to view a larger version of this figure.
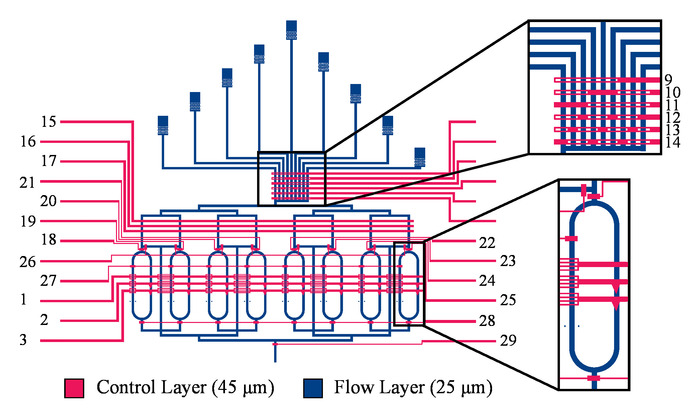
Figure 5. Overview of the microfluidic device design. The microfluidic flow reactor for continuous IVTT reactions consists of eight reactor rings, each with a volume of 10.7 nL. Nine inlets allow for the inflow of nine unique reaction solutions into the device. 24 control channels regulate the flow of fluids within the device. Control channels 9 through 14 form a multiplexer. These control channels should be pressurized at all times to inhibit fluid flow into the device. Depressurisation of two control channels simultaneously allows for the inflow of a single reagent. Control channels 15, 16, and 17 are used to peristaltically pump the reagents into the device in a controlled manner. Control channels 18 through 25 each control the inlet of one of the eight reactors found within the device. Control channel 26 can close the flush channel, thus forcing fluid into the reactors. Control channel 27 aids in the homogeneous filling of the reactors. Control channels 28 and 29 regulate the ring reactor outlets and the only device outlet respectively. Finally, control channels 1, 2, and 3 are used to peristaltically pump the fluid within the ring reactors, resulting in mixing of the reagents. The design of this microfluidic device and the figure are both adapted from Neiderholtmeyer et al.29. Please click here to view a larger version of this figure.

Figure 6. Membrane based valve within the microfluidic device. A) Flow channel within the microfluidic device. Two control channels can be seen in the background. These channels are not pressurized and as such the valves are open (fluid can flow). B) The two control channels intersecting the flow layer channels have been pressurized, closing the valves (i.e., fluid flow is impeded). Upon pressurization of the control channels, the thin PDMS membrane separating the flow and control layer channels is deflected upwards (the control layer lies beneath the flow layer) which closes the flow layer channel. The rounding of the flow layer channel is critical in ensuring that the deflected membrane fully closes the flow channel. Please click here to view a larger version of this figure.
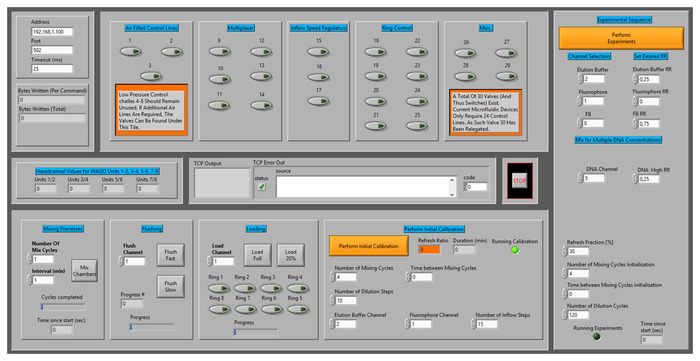
Figure 7. User interface used to control microfluidic device. Throughout this research, a custom control interface has been used to control the flow of fluids within the microfluidic devices. The interface allows users to individually actuate each of the control channels (numbered 1-3 and 9-29), or to execute elaborate protocols resulting in the flushing and loading of reagents, the calibration of the microfluidic device, and the execution of experiments. Please click here to view a larger version of this figure.
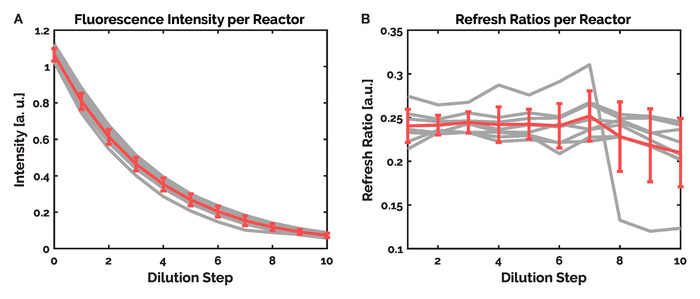
Figure 8. Results of a calibration experiment. During a calibration experiment, the reactors are filled with a fluorophore (25 µM FITC-Dextran) after which the fluorescence intensity is recorded. Subsequently, a series of dilutions follow, where a set number of inflow steps (15) are used to inject ultrapure water into the reactors. After each dilution, the reagents are mixed and the fluorescence is measured. The decrease in the fluorescence intensity per dilution reveals the volume of the reactor ring displaced for the set number of inflow steps; a value termed the Refresh Ratio. A) The average intensity and standard deviation of all eight reactors is shown in red, with the individual intensity traces shown in grey. B) The average Refresh Ratio and standard deviation is shown for each dilution step in red. The individual Refresh Ratios of each individual reactor are shown in grey. It can be seen that seven of the eight reactors show very similar behavior, however one reactor shows fluctuations in the Refresh Ratio after the seventh dilution cycle. This highlights the need for unique Refresh Ratios for each of the reactors, as opposed to using an average Refresh Ratio for the injection of reagents into the reactors. Please click here to view a larger version of this figure.
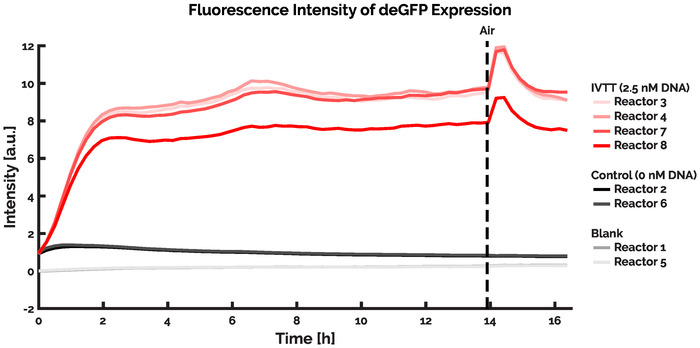
Figure 9. Results of an IVTT experiment expressing the deGFP protein. A prolonged IVTT reaction was initiated such that 30% of the reactor volume is displaced every 14.6 minutes. The reaction was allowed to run for over 16 hours before being terminated. Two reactors of the microfluidic device were used as blanks, with only ultrapure water being flown through the reactors throughout the experiment (reactors 1 and 5). All the other reactors comprised 75% IVTT reaction solution and 25% of either ultrapure water (reactors 2 and 6) or 2.5 nM linear DNA templates coding for the expression of deGFP (reactors 3, 4, 7, and 8). In all four reactors where DNA was added, there is clear deGFP expression. Three of the four reactors provide similar fluorescence intensity, with one reactor displaying lower fluorescence signal. This could be caused by a disparity in flow resulting in less DNA entering the reactor, or due to variations in the reactor dimensions. After 14 hours, a sudden increase is seen in the signal of the reactors containing DNA. This is caused by an air bubble entering the flow layer of the microfluidic device, presumably originating from one of the inflow solutions. The trapping of air in the microfluidic device significantly limits the flow of fluids through the channels, whereby no fresh reagents can be added to or removed from the reactors until the air has passed. Upon resumption of flow, the experiment returns to its previous fluorescence intensity. Please click here to view a larger version of this figure.
Supplemental Files. Please click here to download these files.
