Lentiviral CRISPR/Cas9-Mediated Genome Editing for the Study of Hematopoietic Cells in Disease Models
Summary
Described are protocols for the highly efficient genome editing of murine hematopoietic stem and progenitor cells (HSPC) by the CRISPR/Cas9 system to rapidly develop mouse model systems with hematopoietic system-specific gene modifications.
Abstract
Manipulating genes in hematopoietic stem cells using conventional transgenesis approaches can be time-consuming, expensive, and challenging. Benefiting from advances in genome editing technology and lentivirus-mediated transgene delivery systems, an efficient and economical method is described here that establishes mice in which genes are manipulated specifically in hematopoietic stem cells. Lentiviruses are used to transduce Cas9-expressing lineage-negative bone marrow cells with a guide RNA (gRNA) targeting specific genes and a red fluorescence reporter gene (RFP), then these cells are transplanted into lethally-irradiated C57BL/6 mice. Mice transplanted with lentivirus expressing non-targeting gRNA are used as controls. Engraftment of transduced hematopoietic stem cells are evaluated by flow cytometric analysis of RFP-positive leukocytes of peripheral blood. Using this method, ~90% transduction of myeloid cells and ~70% of lymphoid cells at 4 weeks after transplantation can be achieved. Genomic DNA is isolated from RFP-positive blood cells, and portions of the targeted site DNA are amplified by PCR to validate the genome editing. This protocol provides a high-throughput evaluation of hematopoiesis-regulatory genes and can be extended to a variety of mouse disease models with hematopoietic cell involvement.
Introduction
Many studies in hematology and immunology rely on the availability of genetically modified mice, including conventional and conditional transgenic/knock-out mice that utilize hematopoietic system-specific Cre drivers such as Mx1-Cre, Vav-Cre, and others1,2,3,4,5. These strategies require the establishment of new mouse strains, which can be time-consuming and financially burdening. While revolutionary advances in genome editing technology have enabled the generation of new mouse strains in as few as 3-4 months with the appropriate technical expertise6,7,8,9, much more time is required to amplify the mouse colony before experiments are pursued. In addition, these procedures are costly. For example, Jackson Laboratory lists the current price of knock-out mice generation services at $16,845 per strain (as of December 2018). Thus, methods that are more economical and efficient than conventional murine transgenic approaches are more advantageous.
Clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9 (CRISPR/Cas9) technology has led to the development of new tools for rapid and efficient RNA-based, sequence-specific genome editing. Originally discovered as a bacterial adaptive immune mechanism to destroy invading pathogen DNA, the CRISPR/Cas9 system has been used as a tool to increase the effectiveness of genome editing in eukaryotic cells and animal models. A number of approaches have been employed to transmit CRISPR/Cas9 machinery into hematopoietic stem cells (i.e., electroporation, nucleofection, lipofection, viral delivery, and others).
Here, a lentivirus system is employed to transduce cells due to its ability to effectively infect Cas9-expressing murine hematopoietic stem cells and package together the guide RNA expression construct, promoters, regulatory sequences, and genes that encode fluorescent reporter proteins (i.e., GFP, RFP). Using this method, ex vivo gene editing of mouse hematopoietic stem cells has been achieved, followed by successful reconstitution of bone marrow in lethally irradiated mice10. The lentivirus vector employed for this study expresses the Cas9 and GFP reporter genes from the common core EF1a promoter with an internal ribosomal entry site upstream from the reporter gene. The guide RNA sequence is expressed from a separate U6 promoter. This system is then used to create insertion and deletion mutations in the candidate clonal hematopoiesis driver genes Tet2 and Dnmt3a10. However, the transduction efficiency by this method is relatively low (~5%-10%) due to the large size of the vector insert (13 Kbp) that limits transduction efficiency and reduces virus titer during production.
In other studies, it has been shown that larger viral RNA size negatively affects both virus production and transduction efficiency. For example, a 1 kb increase in insert size is reported to decrease virus production by ~50%, and transduction efficiency will decrease to more than 50% in mouse hematopoietic stem cells11. Thus, it is advantageous to reduce the size of the viral insert as much as possible to improve efficiency of the system.
This shortcoming can be overcome by employing Cas9 transgenic mice, in which the Cas9 protein is expressed in either a constitutive or inducible manner12. The constitutive CRISPR/Cas9 knock-in mice expresses Cas9 endonuclease and EGFP from the CAG promoter at the Rosa26 locus in a ubiquitous manner. Thus, a construct with sgRNA under the control of the U6 promoter and RFP reporter gene under the control of the core EF1a promoter can be delivered using the lentivirus vector to achieve genome editing. With this system, the genes of hematopoietic stem cells have been successfully edited, showing a ~90% transduction efficiency. Thus, this protocol provides a rapid and effective method to create mice in which targeted gene mutations are introduced into the hematopoietic system. While our lab is predominantly using this type of technology to study the role of clonal hematopoiesis in cardiovascular disease processes13,14,15, it is also applicable to studies of hematological malignancy16. Furthermore, this protocol can be extended to the analysis of how DNA mutations in HSPC impact other disease or developmental processes in the hematopoietic system.
To establish a robust lentivirus vector system, high titer viral stocks and optimized conditions for the transduction and transplantation of hematopoietic cells are required. In the protocol, instructions are provided on the preparation of a high titer viral stock in section 1, optimizing the culture conditions of murine hematopoietic stem cells in section 2, methods for bone marrow transplantation in section 3, and assessing engraftment in section 4.
Protocol
All procedures involving animal subjects have been approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Virginia.
1. Generation and purification of lentivirus particles
NOTE: Lentivirus particles containing the optimized guide RNA can be produced by the detailed protocols provided by Addgene: <https://media.addgene.org/cms/files/Zhang_lab_LentiCRISPR_library_protocol.pdf)>. Optimized methods for high-titer lentivirus preparation and storage are discussed elsewhere17,18. In brief, lentiviruses are produced by co-transfection of a lentivirus vector plasmid, psPAX2, and pMD2.G into HEK 293T cells. Culture supernatant is collected at 48 h post-transfection and concentrated by ultracentrifugation. Lentiviral titer is determined by a commercially available qPCR-based assay. This procedure should be performed in a biosafety class II cabinet.
- Prepare a 1:200 solution of collagen (0.0005%) in 1x PBS.
- Coat a 6 well plate with collagen solution and incubate at 37 °C, 5% CO2 for ~30 min.
- Seed 293T cells at a density of 1 x 106 cells per well and incubate at 37 °C, 5% CO2 for ~2 h.
- To prepare the mixture of three transfection plasmids for one well, combine 0.9 µg of lentivirus vector, 0.6 µg of psPAX2, and 0.3 µg of pMD2.G, then achieve a total volume of 10 µL by adding deionized water. Adjust amounts accordingly depending on the number of wells. The amount and ratio of each plasmid may need to be further optimized to suit the researchers needs.
- Carefully add 50 µL of 1x PBS and 5 µL of the diluted PEI MAX (1.0 mg/mL) to the plasmid mixture and incubate for 15 min at room temperature (RT) (Table 1).
- Add 1 mL of DMEM to the mixture.
- Aspirate media from the 6 well plate, add 1 mL of plasmid mixture, and incubate at 37 °C, 5% CO2 for ~3 h.
- Replace the media with 2 mL of fresh DMEM and incubate at 37 °C, 5% CO2 for 24 h.
- Add 1 mL of fresh DMEM and incubate at 37 °C, 5% CO2 for an additional 24 h (total incubation time is 48 h).
- Transfer the culture supernatant to a 50 mL tube and centrifuge at 3,000 x g for 15 min to remove any free-floating cells.
- Filter the supernatant through a 0.45 µm filter.
- Transfer the filtrate to polypropylene centrifuge tubes.
- Ultracentrifuge at 4 °C and 72,100 x g at rmax for 3 h.
- Carefully aspirate the supernatant, leaving behind the white pellet.
- Resuspend the pellet with 100 µL of serum-free hematopoietic cell expansion medium without aeration.
- Keep a 10 µL aliquot to measure the viral titer and store all remaining aliquots at -80 °C until required.
- Titrate the virus with a qPCR-based assay according to the manufacturer's instructions using the 10 µL viral aliquot.
2. Isolation and transduction of lineage-negative cells from mouse bone marrow (Figure 1A)
NOTE: Typically, to isolate enough cells, pairs of tibias, femurs, and humeri are harvested from each mouse. Pelvic and spinal bones may also be harvested as a source of lineage-negative cells.
- Isolation of bone marrow cells
- Euthanize 8-10 week old male CRISPR/Cas9 knock-in mice by 5% isoflurane followed by cervical dislocation, then disinfect their skin with 70% ethanol.
- Using dissecting scissors, make a transverse incision in the skin just below the ribcage and peel the skin distally in both directions to expose the legs and arms.
- Carefully separate the lower limbs from the hip bone by dislocating the hip joint. Cut along the femur head to remove the femur completely from the hip. Dislocate the knee and cut at the joint to separate the femur and tibia, while keeping the bone epiphysis intact. Dislocate the ankle joint and peel away the foot and extra muscle.
- Using dissecting scissors, cut over the shoulder to detach the upper limbs. Dislocate the shoulder, then cut at the elbow joint to harvest the humerus bone.
- Use cellulose-fiber wipes to carefully remove muscles from the femurs, tibias, and humeri. Take extra precaution to ensure that the bones do not break during this process.
- Place the isolated bones into a 50 mL conical tube containing RPMI, and place on ice.
NOTE: The following steps should be carried out in a biosafety class II cabinet. - Transfer the bones into a sterile, 100 mm culture dish.
- Grasp the bone with blunt forceps, and using dissecting scissors, carefully cut both epiphyses.
NOTE: An insufficient cutting will lead to an incomplete flush of bone marrow, while overly aggressive cutting will result in cell loss. - Fill a 10 mL syringe with ice-cold RPMI, and using a 22 G needle, flush the bone marrow from the shaft into a new 100 mm culture dish.
NOTE: Bones will become white and translucent if the bone shaft has been well-flushed. If not, re-cut the bone ends and flush again. - After all the bone marrow has been collected, make a single-cell suspension by passing the bone marrow several times through a 10 mL syringe with an 18 G needle. Repeat 10x to ensure a single-cell suspension.
- Filter cell suspension through a 70 µm cell strainer into a 50 mL conical tube.
- Centrifuge at 310 x g for 10 min at 4 °C.
- Aspirate the supernatant and resuspend the cell pellets in an appropriate volume of optimized separation buffer for the following cell separation process.
- Isolation and lentivirus transduction of lineage-negative cells
NOTE: Mouse lineage-negative cells are isolated from the bone marrow of Cas9 transgenic mice3, or other strains of mice, using a lineage depletion kit according to the manufacturer's instructions. Typically, lineage-negative cells account for 2%-5% of whole bone marrow nucleated cells, and the purity is usually greater than 90% following isolation. The isolated lineage-negative cells are cultured in serum-free hematopoietic cell expansion medium supplemented with 20 ng/mL recombinant murine TPO and 50 ng/mL recombinant murine SCF, then transduced with the lentivirus vector for 16 h at a multiplicity of infection (MOI) = 100.- To isolate lineage-negative cells, use the lineage cell depletion kit according to the manufacturer's instructions.
- After isolation resuspend the lineage-negative cells in 1 mL of serum-free hematopoietic cell expansion medium.
- Seed the cells into a 6 well plate at a density of 1.5 x 106 cells/mL (5 x 105 lineage-negative cells/mouse.)
- Add recombinant murine TPO and SCF into wells at final concentrations of 20 ng/mL and 50 ng/mL, respectively.
- Pre-incubate cells at 37 °C in 5% CO2 for ~2 h.
- Add lentivirus at MOI = 100, 4 µg/mL polybrene, and penicillin/streptomycin to the wells and incubate at 37 °C, 5% CO2 for 16-20 h (Figure 1B).
- On the following day, collect the lentivirus transduced cells into a 15 mL conical tube and centrifuge at 300 g for 10 min.
- Carefully aspirate the supernatant and resuspend the pellet in 200 µL of RPMI per mouse. Keep the cells at RT until transplantation into mice (section 3).
3. Transplantation of transduced cells into lethally irradiated mice
- On the day of bone marrow transplantation, place recipient mice into an eight-slice pie cage and expose them to two doses of whole body irradiation (550 Rad/dose, total dose = 1100 Rad), with approximately 4 h between each irradiation session.
- After the second irradiation session, inject transduced lineage-negative cells to each anesthetized recipient mouse via the retro-orbital vein plexus (200 µL in total) using an insulin syringe (Figure 1C).
- After irradiation, mice should be housed in sterilized cages and provided with a soft diet and drinking water supplemented with antibiotics for 14 d.
- At 3-4 weeks after bone marrow transplantation, analyze peripheral blood to check for the engraftment of transduced donor cells (section 4).
4. Evaluating the chimerism of peripheral blood
- Anesthetize mice with 5% isoflurane and obtain a blood sample from a retro-orbital vein using capillary tubes, and collect it into K2EDTA tubes (the volume in one capillary tube is sufficient for the following assay).
- Transfer 20 µL of blood from the K2EDTA tubes into the 5 mL round bottom polystyrene test tubes, and put on ice.
- Add 1.5 mL of RBC lysis buffer to lyse red blood cells. Incubate for 5 min on ice.
- To neutralize the lysis buffer, wash samples with FACS buffer (1.5 mL/sample).
- Centrifuge at 609 x g at rmax for 5 min at 4 °C. Discard the supernatant.
- Incubate the cells with a cocktail of monoclonal antibodies (diluted in 100 µL FACS buffer/sample) at RT for 20 min in the dark. A complete list of antibodies is provided in the Materials section above.
- Wash the cells once with FACS buffer (2 mL/sample). Centrifuge at 609 x g at rmax (1,800 rpm) for 5 min at 4 °C. Discard the supernatant completely.
- Fix the cells with paraformaldehyde containing fixation buffer (100 µL/tube) for 10 min at 4 °C.
- Wash cells once with FACS buffer (3 mL/sample). Centrifuge at 609 x g at rmax (1,800 rpm) for 5 min at 4 °C. Discard the supernatant completely.
- Suspend the pellet in 400 µL of FACS buffer.
- Keep the samples at 4 °C until analysis by flow cytometry.
Representative Results
Using the above described protocol, approximately 0.8-1.0 x 108 bone marrow cells per mouse have been obtained. The number of lineage-negative cells we obtain is approximately 3 x 106 cells per mouse. Typically, the yield of bone marrow lineage-negative cells is 4%-5% of that of total bone marrow nuclear cells.
Chimerism of transduced cells (RFP-positive) is evaluated by flow cytometry of the peripheral blood (Figure 2A,B). Blood is isolated from the retro-orbital vein and appropriate markers are used to determine the identity of each hematopoietic cell population (i.e., neutrophils, monocytes, T cells, etc.) (Figure 3A,B). Genomic DNA can be isolated from RFP-positive blood cells, and sections of the targeted site DNA can be amplified by PCR and subcloned into TA cloning vectors for sequence analysis. These plasmids are transduced into E. coli and the target site sequences are determined by Sanger sequencing (Figure 4). Alternatively, target site sequences can be determined by other methods, such as Sanger sequencing of the pooled genome followed by tracking of indels by decomposition (TIDE) analysis10. For the control condition, mice are typically transplanted with cells that are transduced with a lentivirus expressing non-targeting guide RNA.
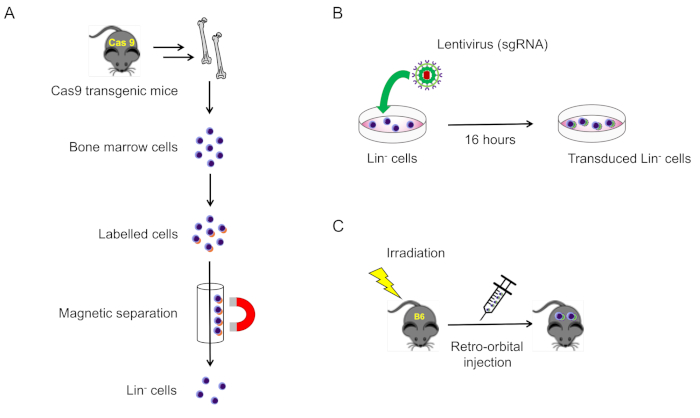
Figure 1: Schematic illustration of this protocol. (A) Isolation of lineage-negative bone marrow cells from Cas9-expressing mice (section 2.1). (B) Lentivirus transduction of lineage-negative cells (section 2.2). (C) Retro-orbital injection of transduced cells into lethally irradiated wild type mice (section 3). Please click here to view a larger version of this figure.
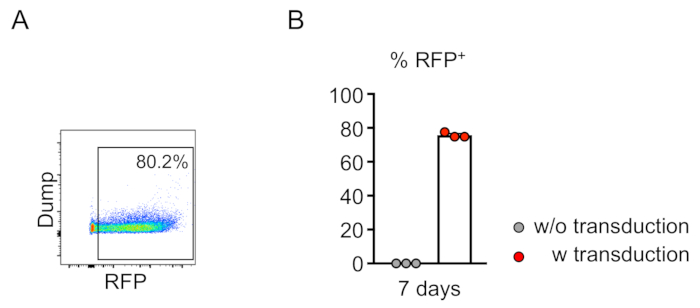
Figure 2: Efficient lentiviral transduction of mouse bone marrow lineage-negative cells in vitro. (A) Flow cytometry analysis reveals successful transduction of lineage-negative cells. Analysis was performed after 7 days of in vitro culture. (B) On average, 75.7% of cells were transduced in this assay (n = 3). Please click here to view a larger version of this figure.
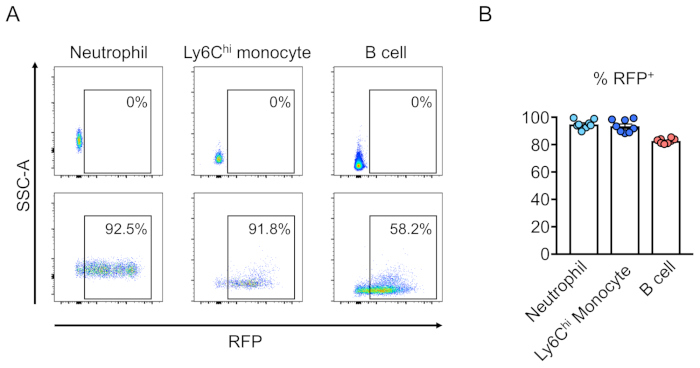
Figure 3: Reconstitution of lethally-irradiated mouse bone marrow by transduced lineage-negative cells.(A) Flow cytometry analysis of mouse peripheral blood following reconstitution by hematopoietic stem cells that were (bottom) or were not (top) transduced with lentivirus expressing RFP. Neutrophils are defined as Ly6G+ and Ly6Chi monocytes as Ly6G– and Ly6C+, and B cells as CD45R+. (B) In these assays, an average of 94.8%, 93.5%, and 82.7% of cells are RFP+ in the neutrophil, Ly6Chi monocyte, and B cell populations, respectively (n = 8). Please click here to view a larger version of this figure.
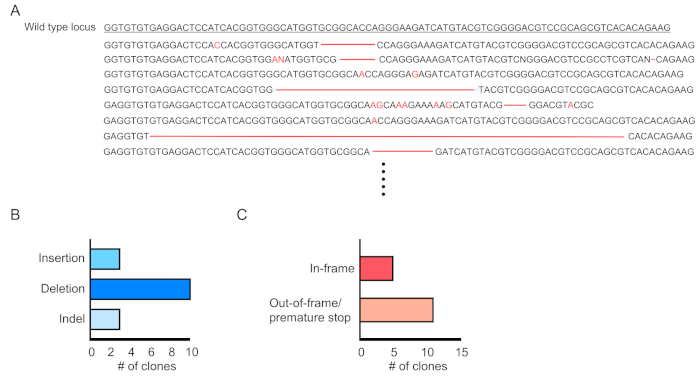
Figure 4: Evaluation of gene editing in transduced blood cells.(A) Example of gene editing showing sequencing results of mutated Dnmt3a locus in RPF-positive blood cells. Deletions are shown as red dashes and insertions are denoted with red letters. (B) Summary of mutations that were detected. (C) 69% (11/16 clones) showed out-of-frame/premature stop mutations. Please click here to view a larger version of this figure.
| Plasmid | Size (bp) | Amount per well (µg) | Ratio |
| pLKO5.0 | 7700 | 0.9 | 2 |
| psPAX2 | 10668 | 0.6 | 1 |
| pMD2.G | 5822 | 0.3 | 1 |
| PEI-max (stock: 100 mg/mL) | 5 µL/well | ||
Table 1: Amounts of plasmid and PEI-max used for transfection.
Discussion
The advantage of this protocol is the creation of animal models harboring specific mutations in hematopoietic cells in a rapid and highly cost-effective manner compared to conventional mouse transgenic approaches. It was found that this methodology enables the generation of mice with hematopoietic cell gene-manipulations within 1 month. There are several critical steps in this protocol that require further consideration.
Screening of gRNA sequence
It is recommended to test gRNAs in vitro to assess editing efficiency prior to conduct in vivo experiments. The efficiency of gRNAs is tested using a cell-free in vitro transcription and screening system. The transcribed gRNA is validated by measuring its efficiency at cleaving the template DNA in the presence of recombinant Cas9 protein, using agarose gel electrophoresis. Commercially available kits are available for this purpose.
Here, indel mutations are characterized by the TA cloning of PCR products amplified from edited region, transforming bacterial cells with those plasmids, and picking up individual colonies for Sanger sequencing. However, this method is laborious and time-consuming. Alternatively, next-generation sequencing (NGS) or pooled DNA sequencing followed by TIDE analysis can be performed19. The TIDE algorithm was created to analyze Sanger sequence traces generated from complex samples. It has been shown that indel estimates with TIDE are typically consistent with those off-targeted NGS20. The analytical software is available online at <http://tide.nki.nl>.
Generation of high-titer lentivirus particles
The viral vesicular stomatitis virus G-protein, which is essential for cell infection, is highly pH-sensitive. Thus, it is important to keep the culture medium within an acceptable pH range, and it should not develop a yellowish appearance. Collagen-coated dishes for virus generation are employed because it accelerates the attachment of HEK293T cells and allows the performance of transfection within a few hours, rather than waiting overnight. However, depending on the experimental schedule, overnight incubation can also be considered.
Purification of lentivirus particles
To achieve efficient transduction of hematopoietic stem cells, it is necessary to generate high-titer lentivirus. Optimization of centrifugation speed is a key feature. While the concentration of lentivirus is usually performed at 90,000 x g, several reports have shown that virus recovery increases if the material is centrifuged at the lower speed of 20,000 x g18. The production of high-titer lentivirus preparation without ultracentrifugation has also been suggested17. It should be noted that it is important to suspend the virus centrifugation pellet while avoiding vigorous pipetting to minimize aeration and maintain virus integrity. High-titer lentivirus particles are required for efficient transduction of hematopoietic stem cells11. Pilot experiments revealed that an MOI of 100 is optimal with regards to transduction efficiency and cell viability. It is recommended to evaluate lentivirus stocks on the basis of cell viability and transduction efficiency.
Storage of lentivirus particles
The lentivirus titer is highly sensitive to temperature, and the titer can be drastically reduced by inappropriate storage conditions and repeated freeze-thaw cycles. It has been found that transduction efficiency of lentivirus decreases rapidly when stored at 4 °C [t(1/2) = 1.3 days] or subjected to multiple freeze-thaw cycles [t(1/2) = 1.1 rounds]. It is recommended that the virus preparations be snap-frozen in liquid nitrogen or crushed dry ice soon after the virus pellet is suspended. The viral stocks should be maintained at -80 °C and thawed on ice to RT just prior equilibration and use11.
Several potential limitations should be noted. First, the introduction of off-target indel mutations by CRISPR/Cas9 has long been appreciated. It has also been shown that CRISPR/Cas9 can induce off-target mutations in vivo21. In practice, off-target indel mutations can be avoided by using gRNA sequences that are closely matched to target genome sites and have more than four mismatches to predicted secondary sites. Such design can be done with existing in silico tools22. Other computational tools to predict gRNA with minimized off-target actions are available (<http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design> or <http://www.Benchling.com>). It may also be beneficial to analyze an animal model using two or more different gRNAs to confirm the phenotype and minimize the possibility that the observed phenotype is mediated by an off-target effect of a specific gRNA.
In addition to conventional indel mutations created by CRISPR/Cas9, larger deletions that extend beyond kilobases have been reported. This can confound studies; however, those larger deletions are reported to be much lower frequency compared to indels23. Another potential problem is genetic compensation. It has been reported that mutant RNA with a premature termination codon (PTC) can result in the upregulation of related genes with sequence similarity by COMPASS complex-mediated activation of transcription24,25. This event has been suggested to be a mechanism that can lead to phenotypic differences between knock-out and knockdown approaches of gene ablation. Because CRISPR/Cas9-mediated genome editing heavily relies on stochastic introduction of frame-shift mutations that lead to the generation of PTC, genetic compensation can modify the phenotype. To avoid genetic compensation, experiments can be considered in which a gene's regulatory sequences are targeted by CRISPR/Cas9 or by the introduction of epigenetic modifiers using Cas9 as an RNA-guided DNA recognition platform.
Lastly, it should be acknowledged that hematopoiesis from cells engrafted into lethally-irradiated mice may differ from the native conditions of hematopoiesis. Furthermore, irradiation can have systemic effects on the organism that may confound interpretation of experiments examining the consequences gene mutations in hematopoietic cells.
Researchers have taken advantage of catalytically inactive Cas9 (dCas9) proteins as a "RNA-guided DNA recognition platform" and used dCas9 fusion proteins to localize effector domains to specific DNA sequences to either repress (CRISPRi) or activate (CRISPRa) transcription off-target genes26,27. While this protocol uses catalytically active Cas9 transgenic mice to introduce dsDNA cleavage in genomic DNA sequence, epigenetic modification to repress or activate specific genes is applicable by fusing dCas9 with chromatin modifier domains such as dCas9-KRAB or dCas9-VP64, respectively. Alternatively, dCas9 can be used as a transcriptional repressor as its own, by blocking transcriptional machinery to access to the gene site27. More recently, Zhou et al. established dCas9-SunTag-p65-HSF1 (SPH) transgenic mice that express a modified version of an epigenetic activator fused with dCas9 and showed that this CRISPRa system is functional in vivo28.
Our lab predominantly uses this technology to study the role of clonal hematopoiesis in cardiovascular disease processes. In proliferating tissue, somatic mutations in cancer driver genes can confer a cellular growth advantage and lead to aberrant clonal expansions. In the hematopoietic system, this process is known as "clonal hematopoiesis", and it results in situations in which a substantial fraction of an individual's leukocytes are replaced by mutant clones. There is a growing appreciation that aberrant clonal expansions accelerate cardiovascular disease, such as atherosclerosis and heart failure, and contribute to morbidity and all-cause mortality15,29.
Recently, a causal connection between several of these somatic mutations and cardiovascular disease has been documented, and aspects of the underlying mechanisms have been elucidated10,13,14. However, these somatic mutations probably represent the "tip of the iceberg", as epidemiological studies have shown that many additional candidate genes are associated with clonal hematopoiesis and, potentially, increased cardiovascular disease mortality. Thus, a systematic, higher throughput evaluation of clonal hematopoiesis driver genes is required. Current studies of the causal connection of clonal hematopoiesis and cardiovascular disease are based on the analysis of mice with hematopoietic system-specific conditional transgenic (Mx1-Cre, Vav-Cre, etc.) or mice after bone marrow transplant. These strategies, however, need to establish new mouse colonies and may become a financial and physical burden for researchers. Thus, a cheaper and more rapid method than the conventional murine transgenic/knock-out approach employed in the past is warranted. Lentiviral vectors to transduce HSPC and CRISPR technologies to engineer mutations, as described in this manuscript, facilitate the study of clonal hematopoiesis and cardiovascular disease.
In addition to generating conventional knock-out locus, this method is applicable to the production of truncated mutated proteins. For example, researchers have successfully generated a hematopoietic-Ppm1d truncation, which is frequently seen in patients with clonal hematopoiesis, by introducing frameshift mutations with a gRNA targeting exon 6 of the Ppm1d gene30.
Declarações
The authors have nothing to disclose.
Acknowledgements
S. S. was supported by an American Heart Association postdoctoral fellowship 17POST33670076. K. W. was supported by NIH grants R01 HL138014, R01 HL141256, and R01 HL139819.
Materials
| 1/2 cc LO-DOSE INSULIN SYRINGE | EXELINT | 26028 | general supply |
| 293T cells | ATCC | CRL-3216– | Cell line |
| APC-anti-mouse Ly6C (Clone AL-21) | BD Biosciences | 560599 | Antibodies |
| APC-Cy7-anti-mouse CD45R (RA3-6B2) | BD Biosciences | 552094 | Antibodies |
| BD Luer-Lok disposable syringes, 10 ml | BD | 309604 | general supply |
| BD Microtainer blood collection tubes, K2EDTA added | BD Bioscience | 365974 | general supply |
| BD Precisionglide needle, 18 G | BD | 305195 | general supply |
| BD Precisionglide needle, 22 G | BD | 305155 | general supply |
| BV510-anti-mouse CD8a (Clone 53-6.7) | Biolegend | 100752 | Antibodies |
| BV711-anti-mouse CD3e (Clone 145-2C11) | Biolegend | 100349 | Antibodies |
| Collagen from calf skin | Sigma-Aldrich | 9007-34-5 | general supply |
| Corning Costar Ultra-Low Attachment Multiple Well Plate, 6 well | Millipore Sigma | CLS3471 | general supply |
| CRISPR/Cas9 knock-in mice | The Jackson Laboratory | 028555 | mouse |
| DietGel 76A | Clear H2O | 70-01-5022 | general supply |
| Dulbecco’s Modified Eagle’s Medium (DMEM) – high glucose | Sigma Aldrich | D6429 | Medium |
| eBioscience 1X RBC Lysis Buffer | Thermo fisher Scientific | 00-4333-57 | Solution |
| Falcon 100 mm TC-Treated Cell Culture Dish | Life Sciences | 353003 | general supply |
| Falcon 5 mL round bottom polystyrene test tube | Life Sciences | 352054 | general supply |
| Falcon 50 mL Conical Centrifuge Tubes | Fisher Scientific | 352098 | general supply |
| Falcon 6 Well Clear Flat Bottom TC-Treated Multiwell Cell Culture Plate | Life Science | 353046 | general supply |
| Fisherbrand microhematocrit capillary tubes | Thermo Fisher Scientific | 22-362566 | general supply |
| Fisherbrand sterile cell strainers, 70 μm | Fisher Scientific | 22363548 | general supply |
| FITC-anti-mouse CD4 (Clone RM4-5) | Invitrogen | 11-0042-85 | Antibodies |
| Fixation Buffer | BD Bioscience | 554655 | Solution |
| Guide-it Compete sgRNA Screening Systems | Clontech | 632636 | Kit |
| Isothesia (Isoflurane) solution | Henry Schein | 29404 | Solution |
| Lenti-X qRT-PCR Titration Kit | Takara | 631235 | Kit |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Kit |
| Millex-HV Syringe Filter Unit, 0.45 mm | Millipore Sigma | SLHV004SL | general supply |
| PBS pH7.4 (1X) | Gibco | 10010023 | Solution |
| PE-Cy7-anti-mouse CD115 (Clone AFS98) | eBioscience | 25-1152-82 | Antibodies |
| PEI MAX | Polysciences | 24765-1 | Solution |
| Penicillin-Streptomycin Mixture | Lonza | 17-602F | Solution |
| PerCP-Cy5.5-anti-mouse Ly6G (Clone 1A8) | BD Biosciences | 560602 | Antibodies |
| pLKO5.sgRNA.EFS.tRFP | Addgene | 57823 | Plasmid |
| pMG2D | Addgene | 12259 | Plasmid |
| Polybrene Infection/Transfection Reagent | Sigma Aldrich | TR-1003-G | Solution |
| Polypropylene Centrifuge Tubes | BECKMAN COULTER | 326823 | general supply |
| psPAX2 | Addgene | 12260 | Plasmid |
| RadDisk – Rodent Irradiator Disk | Braintree Scientific | IRD-P M | general supply |
| Recombinant Murine SCF | Peprotech | 250-03 | Solution |
| Recombinant Murine TPO | Peprotech | 315-14 | Solution |
| StemSpan SFEM | STEMCELL Technologies | 09600 | Solution |
| TOPO TA cloning kit for sequencing with One Shot TOP10 Chemically Competent E.coli | Thermo fisher Scientific | K457501 | Kit |
| Zombie Aqua Fixable Viability Kit | BioLegend | 423102 | Solution |
Referências
- Sasaki, Y., et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. EMBO Journal. 32, 2463-2476 (2013).
- Ogilvy, S., et al. Transcriptional regulation of vav, a gene expressed throughout the hematopoietic compartment. Blood. 91, 419-430 (1998).
- Meyer, S. E., et al. DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer Discovery. 6, 501-515 (2016).
- Kuhn, R., Schwenk, F., Aguet, M., Rajewsky, K. Inducible gene targeting in mice. Science. 269, 1427-1429 (1995).
- Cole, C. B., et al. PML-RARA requires DNA methyltransferase 3A to initiate acute promyelocytic leukemia. Journal of Clinical Investigation. 126, 85-98 (2016).
- Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154, 1370-1379 (2013).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153, 910-918 (2013).
- Quadros, R. M., et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biology. 18, 92 (2017).
- Miura, H., Quadros, R. M., Gurumurthy, C. B., Ohtsuka, M. Easi-CRISPR for creating knock-in and conditional knock-out mouse models using long ssDNA donors. Nature Protocols. 13, 195-215 (2018).
- Sano, S., et al. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circulation Research. 123, 335-341 (2018).
- Cante-Barrett, K., et al. Lentiviral gene transfer into human and murine hematopoietic stem cells: size matters. BMC Research Notes. 9 (312), (2016).
- Chu, V. T., et al. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proceedings of the National Academy of Sciences of the United States of America. 113, 12514-12519 (2016).
- Fuster, J. J., et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 355, 842-847 (2017).
- Sano, S., et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. Journal of the American College of Cardiology. 71, 875-886 (2018).
- Sano, S., Wang, Y., Walsh, K. Clonal Hematopoiesis and Its Impact on Cardiovascular Disease. Circulatory Journal. 71, 875-886 (2018).
- Heckl, D., et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nature Biotechnology. 32, 941-946 (2014).
- Jiang, W., et al. An optimized method for high-titer lentivirus preparations without ultracentrifugation. Scientific Reports. 5, 13875 (2015).
- Cribbs, A. P., Kennedy, A., Gregory, B., Brennan, F. M. Simplified production and concentration of lentiviral vectors to achieve high transduction in primary human T cells. BMC Biotechnology. 13, 98 (2013).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42, e168 (2014).
- Sentmanat, M. F., Peters, S. T., Florian, C. P., Connelly, J. P., Pruett-Miller, S. M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Scientific Reports. 8, 888 (2018).
- Akcakaya, P., et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 561, 416-419 (2018).
- Bae, S., Park, J., Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 30, 1473-1475 (2014).
- Kosicki, M., Tomberg, K., Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology. 36, 765-771 (2018).
- El-Brolosy, M. A., et al. Genetic compensation triggered by mutant mRNA degradation. Nature. 568, 193-197 (2019).
- Ma, Z., et al. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature. 568, 259-263 (2019).
- Gilbert, L. A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 154, 442-451 (2013).
- Qi, L. S., et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 152, 1173-1183 (2013).
- Zhou, H., et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nature Neuroscience. 21, 440-446 (2018).
- Fuster, J. J., Walsh, K. Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circulatory Research. 122, 523-532 (2018).
- Kahn, J. D., et al. PPM1D truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood. , (2018).
