Diagnosis
Key features of SSc-ILD on HRCT commonly include a non-specific interstitial pneumonia (NSIP) pattern with peripheral ground-glass opacities and extensive traction bronchiectasis (Figure 1 and Figure 2). Ground-glass opacities have a broad etiology and are often non-specific40,41,42. Central predominance or peripheral distribution with subpleural sparing is highly suggestive of NSIP (Figure 3).
Typically, ILD patterns in HRCT images include reticulations with architectural distortion resulting in traction bronchiectasis/bronchiolectasis (consistent with a fibrotic form of NSIP). Indeed traction bronchiectasis and traction bronchiolectasis are often the predominant features of SSc-ILD (Figure 4)43. Additional findings may include honeycombing (Figure 5; more common in limited forms of SSc), interlobular septal thickening and intralobular lines, and micronodules40,44. Honeycombing refers to clustered cystic airspaces of typically consistent diameter (~3–10 mm) with thick, well defined walls31. Honeycombing and traction bronchiectasis are key features of usual interstitial pneumonia (UIP) on HRCT. Although this pattern is most commonly associated with idiopathic pulmonary fibrosis (IPF), the prototype fibrosing ILD with a progressive phenotype, it can sometimes be seen in patients with SSc-ILD10. Recently, several signs have been identified in patients with connective tissue disease-related ILD (including SSc-ILD) and the UIP pattern on HRCT, but not in those with IPF. These are the straight edge sign (i.e., isolation of fibrosis to the lung bases with sharp demarcation in the craniocaudal plane without substantial extension along the lateral margins of the lungs on coronal images), the honeycombing predominant (or exuberant) sign (>70% of fibrotic portions of the lung), and the anterior upper lobe sign (i.e., concentration of fibrosis within the anterior aspect of the upper lobes, with relative sparing of the other aspects of the upper lobes, and concomitant lower lobe involvement)45. The straight edge sign has also been associated with NSIP pathology46, which is the main CT pattern in SSc-ILD10.
Dilated air-filled esophagus is frequently observed in patients with SSc (Figure 6)47,48,49 and in patients with SSc-ILD47,48. While there is no accepted upper age limit where a dilated esophagus may no longer help to differentiate SSc-ILD and IPF, a dilated esophagus may be more difficult to interpret in patients over age 65 due to increasing incidence of esophageal motility disorders. Mediastinal lymphadenopathy (usually reactive), in which the short axis of the lymph node exceeds 10 mm, is also often observed in patients with SSc-ILD47,50. Pulmonary artery size greater than the adjacent ascending aorta suggests coexistent pulmonary hypertension (Figure 6), even in patients without fibrotic lung disease51,52,53. Areas of consolidation suggest superimposed infection, aspiration, organizing pneumonia, hemorrhage or malignancy. Nodules must be monitored due to the increased risk for lung cancer in SSc-ILD7; the most common primary cancer to arise in patients with SSc-ILD is adenocarcinoma7,54.
SSc-ILD shares a number of clinical, mechanistic, and pathological similarities with IPF15,55. However, some radiologic features allow the differentiation of these two ILDs15,45. In SSc-ILD, compared with IPF, there is a greater proportion of ground-glass opacity and fibrosis is less coarse. In cases of UIP in SSc, honeycombing may be observed in more than 70% of the fibrotic-lung tissue ─ the exuberant honeycombing sign56,57. In addition, the four-corners sign (also known as the anterior upper lobe sign) is significantly more common in SSc-ILD than in IPF; this is a pattern of inflammation and/or fibrosis focally or disproportionately involving the bilateral anterolateral upper lobes and posterosuperior lower lobes58.
Chest radiographs may initially detect ILD; however, they do not offer enough contrast resolution for reliable diagnosis. In chest radiographs from patients with SSc-ILD, the most frequent pattern is basal predominant reticulation59. Further features may include visible bronchiectasis, volume loss and honeycombing. As with HRCT, the presence of a dilated air-filled esophagus may be helpful in supporting the diagnosis of SSc-ILD47.
Prognosis
Several different imaging findings have been shown to be associated with prognosis in SSc-ILD. Mortality risk has been reported to be higher in patients with a disease extent of at least 20% on HRCT (10-year survival was 43% versus 67%, respectively, in patients with disease extent above versus below the 20% threshold)60. Similarly, a high fibrosis score on HRCT (based on the extent of reticulation and honeycombing) has been associated with increased mortality61. Large esophageal diameters are associated with increased ILD severity and decreased DLCO48. Lung density and pulmonary artery diameter may potentially be used to predict the risk of pulmonary hypertension62. Computerized, quantitative CT parameters could also be harnessed to identify patients’ risk of lung function decline or mortality. One study suggested that the extent of ILD, quantified from HRCT, could be used to predict the decline in FVC over 12 months63. In another study, quantitative chest CT parameters provided mortality risk results that were consistent with clinical prediction models64. Despite their apparent potential, imaging-based biomarkers are currently best considered at a population level as their clinical utility in individual patients has not been established.
Treatment response
Cyclophosphamide and mycophenolate mofetil provide modest benefit in patients with SSc-ILD. In the landmark Scleroderma Lung Study I, cyclophosphamide treatment led to slower progression of fibrosis compared with placebo65. More recently, the Scleroderma Lung Study II reported similar efficacy and improved tolerability with mycophenolate mofetil in comparison with cyclophosphamide66. Based upon positive SENSCIS® trial results67, nintedanib became the first US Food and Drug Administration-approved treatment to slow the rate of decline in lung function in patients with SSc-ILD. However, there remains a need for improved treatment options for patients with SSc-ILD. Therapies currently being investigated include monoclonal antibodies (e.g. rituximab, abituzumab), antifibrotic agents (e.g., pirfenidone), the direct thrombin inhibitor dabigatran, the proteasome inhibitor bortezomib, and hematopoietic stem cell transplantation19,68.
Serial HRCT scans showing disease progression in a patient with SSc-ILD
HRCT assessments performed at different timepoints may be used to investigate disease progression. Figure 7 shows two sets of axial and coronal chest HRCT images taken 10 years apart in a patient with SSc-ILD. The initial axial and coronal images (Figure 7A and B) from chest HRCT show basilar predominant ground-glass opacity and reticulation with mild traction bronchiectasis and subpleural sparing consistent with NSIP in this patient with SSc. The latter set of images (Figure 7C and D) taken 10 years later, show increased reticulation and traction bronchiolectasis at the lung bases with decrease in ground-glass opacity on axial and coronal (Figure 7C and D) images from chest CT consistent with mild worsening of pulmonary fibrosis. Serial HRCT scans can also be used to monitor treatment response69,70,71; this was demonstrated in the Scleroderma Lung Study II, in which computer-aided diagnosis scores based on HRCT scans were used to compare the efficacy of cyclophosphamide with mycophenolate mofetil in patients with SSc-ILD69.
| Phase | Detector collimation |
Voltage (kVp) |
Current (mAs) | Scan interval |
Pitch | Rotation | Tube current modulation |
| Supine inspiratory | Helical 1.2 mm | 120 (may be lowered) | 230 (may be lowered) | N/A | ~1.0 | 0.5 seconds or faster | On |
| Supine expiratory | Axial 2 x 1.0 mm | 120 | 150 | 20 mm | N/A | 1.0 seconds | On |
| Prone inspiratory | Axial 2 x 1.0 mm | 120 | 150 | 20 mm | N/A | 1.0 seconds | On |
Table 1: Computed tomography acquisition parameters37. kVp = kilovoltage peak; N/A = not applicable.
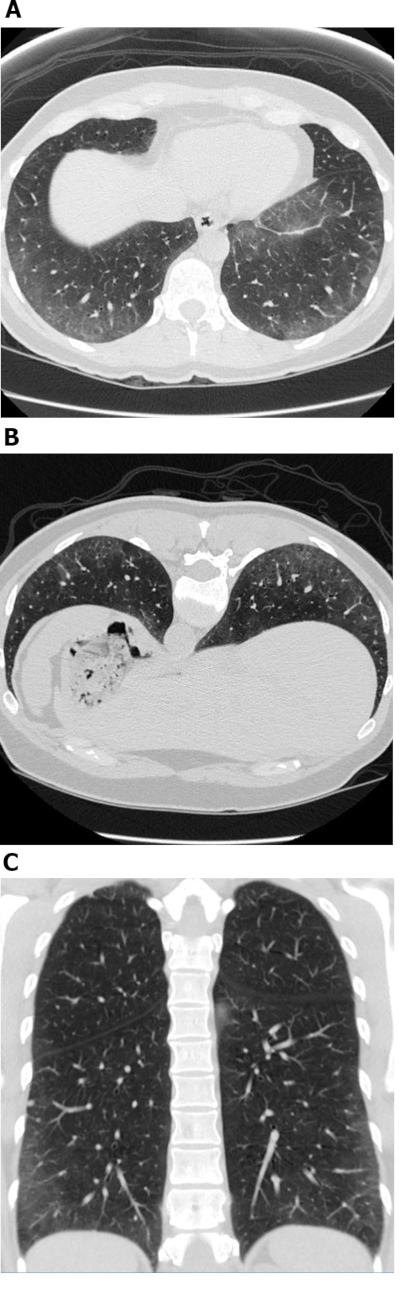
Figure 1: Systemic sclerosis with a cellular NSIP pattern of disease. Axial (A), prone (B) and coronal (C) high-resolution computed tomography images all show extensive peripheral and basal predominant ground-glass opacities; these are typical observations with NSIP. The lack of traction bronchiectasis is suggestive of a cellular NSIP pattern of disease. NSIP = non-specific interstitial pneumonia. Please click here to view a larger version of this figure.
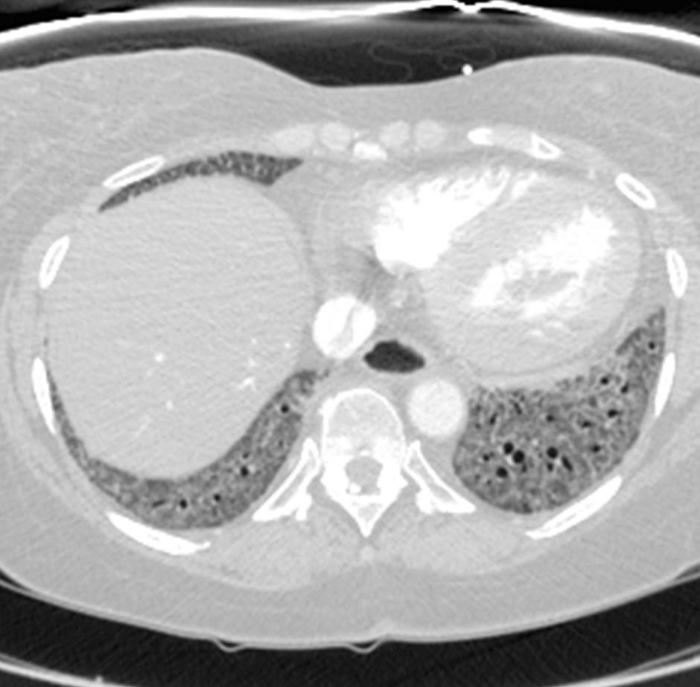
Figure 2: Systemic sclerosis with a fibrotic non-specific interstitial pneumonia pattern of disease. Axial computed tomography image shows extensive, basal-predominant ground-glass opacities with associated traction bronchiectasis. Notably, the esophagus shows marked dilation; this is typical of scleroderma. Please click here to view a larger version of this figure.

Figure 3: Systemic sclerosis with a fibrotic NSIP pattern. Axial high-resolution computed tomography images (A and B) show extensive ground-glass opacities, reticulation, architectural distortion and traction bronchiectasis. Notably, subpleural sparing is apparent; this is typical of NSIP and is seen in about 50% of all cases. NSIP = non-specific interstitial pneumonia. Please click here to view a larger version of this figure.
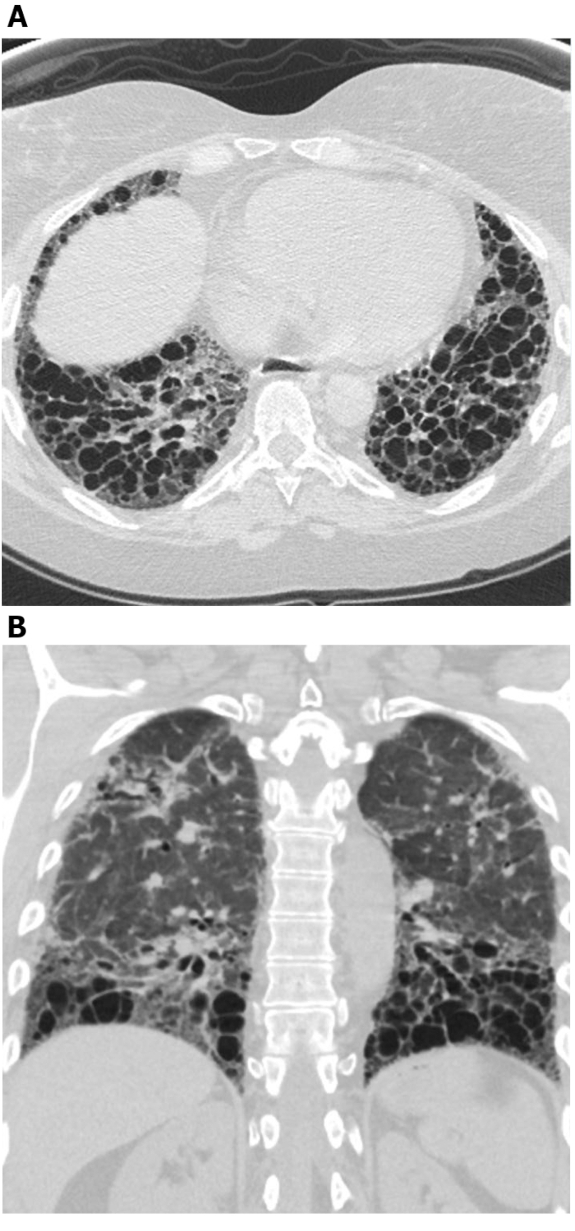
Figure 4: Systemic sclerosis with exuberant traction bronchiectasis. Axial (A) and coronal (B) high-resolution computed tomography images show extensive middle and lower lung zone predominant traction bronchiectasis. While this may be mistaken for honeycombing, the cystic areas connect with each other and spare the immediate subpleural lung; this is typical of bronchiectasis. Please click here to view a larger version of this figure.
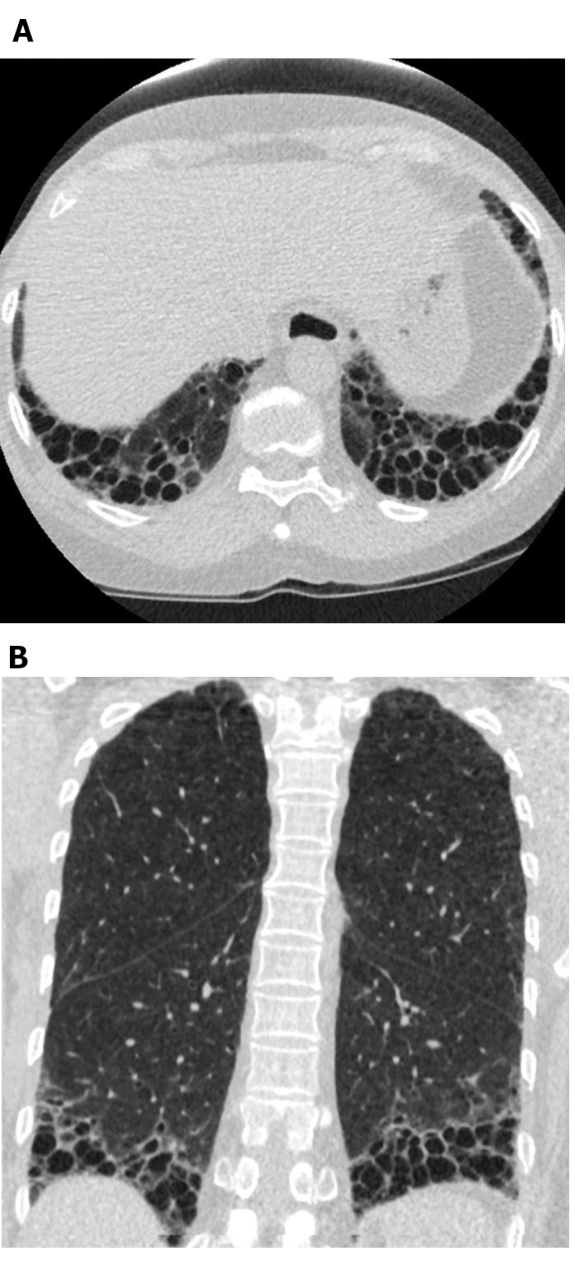
Figure 5: Systemic sclerosis with a UIP pattern of lung fibrosis. Axial (A) and coronal (B) computed tomography images show peripheral and basal predominant honeycombing and traction bronchiectasis in keeping with the typical UIP pattern of lung fibrosis. Note the dilated esophagus (attributable to scleroderma) and the ‘exuberant’ honeycombing (suggestive of ILD related to connective tissue disease rather than idiopathic pulmonary fibrosis). UIP = usual interstitial pneumonia. Please click here to view a larger version of this figure.
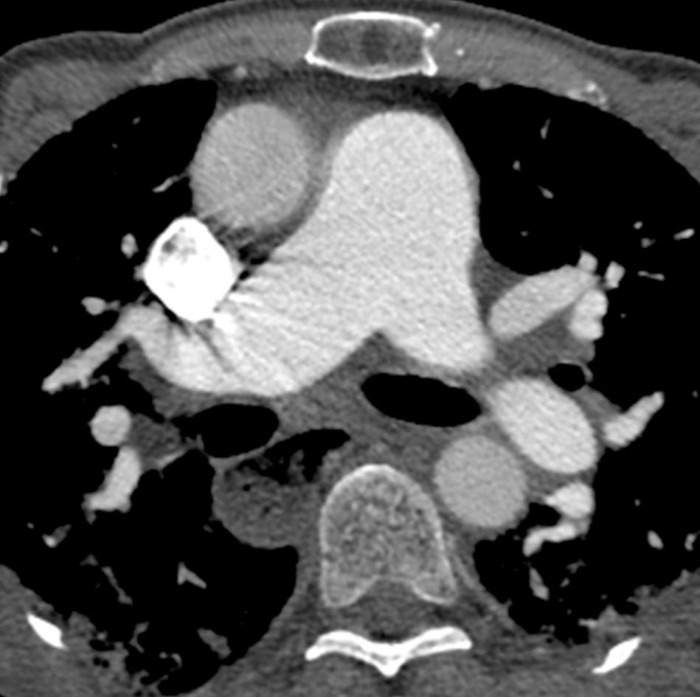
Figure 6: Systemic sclerosis with pulmonary hypertension and dilated esophagus. Contrast-enhanced chest computed tomography shows marked enlargement of the pulmonary trunk, with a larger measurement than the adjacent ascending aorta that suggests underlying pulmonary hypertension. The esophagus is markedly dilated; this is attributable to scleroderma. Please click here to view a larger version of this figure.
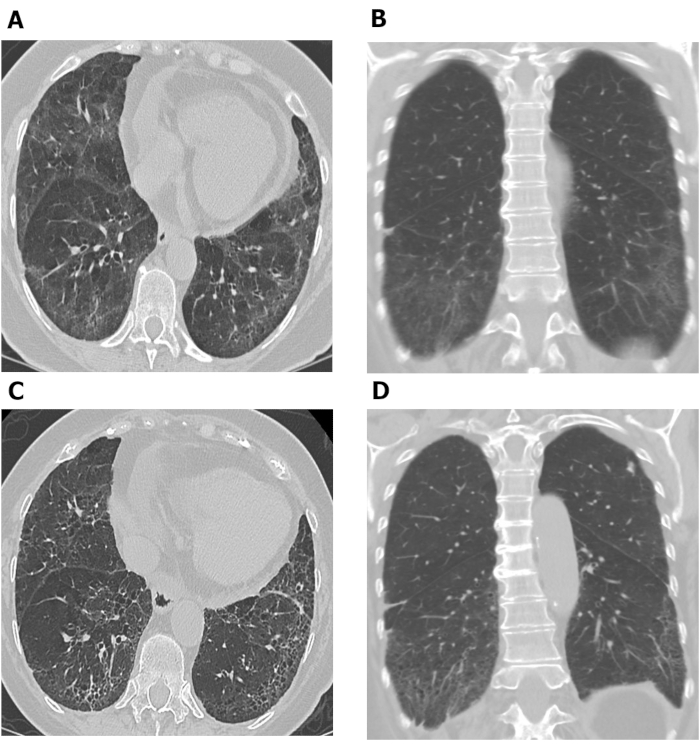
Figure 7: Serial chest HRCT images showing progression of pulmonary fibrosis in patient with SSc-ILD. Axial (A) and coronal (B) images from chest HRCT show basilar predominant ground-glass opacity and reticulation with mild traction bronchiectasis and subpleural sparing consistent with non-specific interstitial pneumonia in this patient with SSc. After 10 years, increased reticulation and traction bronchiolectasis at the lung bases with decrease in ground-glass opacity are observed on axial (C) and coronal (D) chest HRCT images, consistent with mild worsening of pulmonary fibrosis. HRCT = high-resolution computed tomography; SSc-ILD = systemic scleroderma-associated interstitial lung disease. Please click here to view a larger version of this figure.
