Ultrafast Time-resolved Near-IR Stimulated Raman Measurements of Functional π-conjugate Systems
Summary
Details of signal generation and optimization, measurement, data acquisition, and data handling for a femtosecond time-resolved near-IR stimulated Raman spectrometer are described. A near infrared stimulated Raman study on the excited-state dynamics of β-carotene in toluene is shown as a representative application.
Abstract
Femtosecond time-resolved stimulated Raman spectroscopy is a promising method of observing the structural dynamics of short-lived transients with near infrared (near-IR) transitions, because it can overcome the low sensitivity of spontaneous Raman spectrometers in the near-IR region. Here, we describe technical details of a femtosecond time-resolved near-IR multiplex stimulated Raman spectrometer that we have recently developed. A description of signal generation and optimization, measurement, data acquisition, and calibration and correction of recorded data is provided as well. We present an application of our spectrometer to analyze the excited-state dynamics of β-carotene in toluene solution. A C=C stretch band of β-carotene in the second lowest excited singlet (S2) state and the lowest excited singlet (S1) state is clearly observed in the recorded time-resolved stimulated Raman spectra. The femtosecond time-resolved near-IR stimulated Raman spectrometer is applicable to the structural dynamics of π-conjugate systems from simple molecules to complex materials.
Introduction
Raman spectroscopy is a powerful and versatile tool for investigating the structures of molecules in a wide variety of samples from simple gases, liquids, and solids to functional materials and biological systems. Raman scattering is significantly enhanced when the photon energy of the excitation light coincides with the electronic transition energy of a molecule. The resonance Raman effect enables us to selectively observe the Raman spectrum of a species in a sample composed of many kinds of molecules. Near-IR electronic transitions are drawing a lot of attention as a probe for investigating the excited-state dynamics of molecules with large π-conjugated structures. The energy and lifetime of the lowest excited singlet state have been determined for several carotenoids, which have a long one-dimensional polyene chain1,2,3. The dynamics of neutral and charged excitations have been extensively investigated for various photoconductive polymers in films4,5,6,7, nanoparticles8, and solutions9,10,11. Detailed information on the structures of the transients will be obtainable if time-resolved near-IR Raman spectroscopy is applied to these systems. Only a few studies, however, have been reported on time-resolved near-IR Raman spectroscopy12,13,14,15,16, because the sensitivity of near-IR Raman spectrometers is extremely low. The low sensitivity principally originates from the low probability of near-IR Raman scattering. The probability of spontaneous Raman scattering is proportional to ωiωs3, where ωi and ωs are the frequencies of the excitation light and the Raman scattering light, respectively. In addition, commercially available near-IR detectors have much lower sensitivity than CCD detectors functioning in the UV and visible regions.
Femtosecond time-resolved stimulated Raman spectroscopy has emerged as a new method of observing time-dependent changes of Raman active vibrational bands beyond the apparent Fourier-transform limit of a laser pulse17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33. Stimulated Raman scattering is generated by irradiation of two laser pulses: the Raman pump and probe pulses. Here it is assumed that the Raman pump pulse has a larger frequency than the probe pulse. When the difference between the frequencies of the Raman pump and probe pulses coincides with the frequency of a Raman active molecular vibration, the vibration is coherently excited for a large number of molecules in the irradiated volume. Nonlinear polarization induced by the coherent molecular vibration enhances the electric field of the probe pulse. This technique is particularly powerful for near-IR Raman spectroscopy, because stimulated Raman scattering can solve the problem of the sensitivity of time-resolved near-IR spontaneous Raman spectrometers. Stimulated Raman scattering is detected as intensity changes of the probe pulse. Even if a near-IR detector has a low sensitivity, stimulated Raman scattering will be detected when the probe intensity is sufficiently increased. The probability of stimulated Raman scattering is proportional to ωRPωSRS, where ωRP and ωSRS are the frequencies of the Raman pump pulse and stimulated Raman scattering, respectively20. The frequencies for stimulated Raman scattering, ωRP and ωSRS, are equivalent to ωi and ωs for spontaneous Raman scattering, respectively. We have recently developed a femtosecond time-resolved near-IR Raman spectrometer using stimulated Raman scattering for investigating the structures and dynamics of short-lived transients photogenerated in π-conjugate systems2,3,7,10. In this article, we present the technical details of our femtosecond time-resolved near-IR multiplex stimulated Raman spectrometer. Optical alignment, acquisition of time-resolved stimulated Raman spectra, and calibration and correction of recorded spectra are described. The excited-state dynamics of β-carotene in toluene solution is studied as a representative application of the spectrometer.
Protocol
1. Startup of electric devices
- Turn on the femtosecond Ti:sapphire laser system according to its operation manual. Wait 2 h for the laser system to warm up.
- Turn on the power switches of the optical chopper, the translational stage controllers, the spectrograph, the InGaAs array detector, and the computer while the system is warming up. Fill the detector's Dewar with liquid nitrogen.
2. Optical alignment of spectrometer
- Mirror adjustment (Figure 1B)
- Check the position of the support on the mirror mount.
- Turn the upper knob of the mount clockwise and counterclockwise to let the reflected laser beam travel down and up in the vertical direction, respectively, if the support is located at the lower part of the mount. Turn the knob in the opposite direction if the support is located at the upper part of the mount.
- Turn the knob on the left side of the mount clockwise and counterclockwise to let the reflected laser beam travel right and left in the horizontal direction, respectively, if the support is located on the right side of the mount. Turn the knob in the opposite direction if the support is located at the left side of the mount.
- Lens alignment
- Place a business card with a grid behind the lens as a screen.
- Remove the lens. Introduce the incident beam and let it hit the screen. Mark the position of the beam spot on the screen with a pen.
- Block the beam and place the lens. Introduce the beam and confirm that it hits the mark on the screen exactly. If it does not, adjust the vertical and horizontal positions of the lens.
- Prepare a business card with a hole. Let the incident beam pass through the hole in front of the lens and confirm that the specular reflection of the beam by the lens travels in the direction exactly opposite to the incident beam. If it does not, adjust the angle of the lens.
- Laser beam alignment (Figure 1C)
- Place a business card behind iris 2 (i2) as a screen.
- Let the beam pass through the center of i1 by adjusting mirror 1 (m1) according to section 2.1. Let the beam pass through the center of i2 by adjusting m2 according to section 2.1.
- Confirm that the beam passes through the centers of i1 and i2 simultaneously. If the beam does not pass through the center of i1, repeat step 2.3.2 until the beam passes through the centers of both irises.
- Optical delay line alignment (Figure 1D)
- Remove m3 and m4 on the optical delay line (ODL). Place i1 at the position of m3 at the height of the center of m3.
- Move the stage toward m2 as far as it can by placing the direction button of the stage controller. Let the beam pass through the center of i1 by adjusting m1 according to section 2.1.
- Move the stage apart from m2 as far as it can by placing the direction button of the stage controller. Let the beam pass through the center of i1 by adjusting m2 according to section 2.1.
- Move the stage toward the beam input as far as it can and confirm that the beam passes through the center of i1. If the beam does not pass through the center of i1 after step 2.4.3, repeat steps 2.4.2–2.4.3 until the beam passes through the center of i1 at both ends of the stage.
- Remove i1 from the position of m3. Place m3 and m4 on ODL. Let the beam pass through the center of i2 by adjusting m3 and m4 according to steps 2.4.2–2.4.4.
- Once steps 2.4.1–2.4.5 are finished, let the beam pass through the center of i2 by adjusting m1 and m2 according to steps 2.4.2–2.4.5.
- White light continuum generation (Figure 1A)
- Place the variable neutral density filter (VND) VND1 in the incident beam path. Place a business card ~200 mm apart from VND1 as a screen.
- Turn VND1 until the incident beam hits the highest optical density position of VND1, where the transmitted beam has the lowest power.
- Place the lens (L) L1 (focal length = 100 mm) behind VND1. Place the 3 mm thick sapphire plate (SP) ~105 mm apart from L1, where SP is located slightly behind the focus of the beam, letting the beam pass through SP near the edge.
- Set the diameter of I6 to be ~5 mm.
- Turn VND1 to gradually increase the power of the transmitted beam until a yellow-white spot is observed on the screen. Turn VND1 further in the same direction very carefully until a purple ring surrounds the yellow-white spot on the screen.
- Probe beam alignment (Figure 1A)
- Adjust the two pairs of mirrors (M) (M4, M5) and (M7, M8) according to section 2.3. Adjust ODL2 according to section 2.4. Adjust M12 and M13 according to section 2.3.
- Generate a white light continuum according to section 2.5.
- Remove the color glass filters (F) F1 and F2 and the polarizer (P) P1.
- Reflect the white light continuum with the concave mirror (CM). Let the reflected beam pass just beside SP.
- Let the beam hit the center of M15 and M16 by adjusting M14 and M15, respectively, according to section 2.1. Remove L2, L3, and L4. Let the beam hit the center of the entrance slit of the spectrograph by adjusting M16.
- Measure the diameter of the white light continuum beam at CM and the entrance slit using grid paper. If the diameters are significantly altered between the two positions, adjust the position of CM parallel with the beam using a micrometer on the base plate of CM until the diameters become almost identical. Conduct steps 2.6.4–2.6.5 after the adjustment.
- Place L2, L3, and L4 according to section 2.2 and then place F1, F2, and P1.
- Raman pump beam alignment (Figure 1A)
- Place the volume-grating reflective bandpass filter (BPF) in the output beam path of the optical parametric amplifier (OPA) OPA1. Adjust BPF and M17 according to section 2.3. Use a near-IR sensor card for observing the beam spot.
- Set the angle of the half-wave plate (HWP) HWP2 at 45° in order to set the Raman pump polarization to vertical. Remove L5, L6, and L7.
- Let the beam hit the center of M19, M20, and M21 by adjusting M18, M19, and M20, respectively, according to section 2.1. Use a near-IR sensor card to observe the beam spot.
- Place L5, L6, and L7 according to section 2.2 using a near-IR sensor card as a screen.
- Actinic pump beam alignment (Figure 1A)
- Remove L8 and L9. Let the output beam from OPA2 pass through the center of the iris (I) I12 by adjusting M22 according to section 2.1.
- Adjust M24 and M25 according to section 2.3. Place L8 and L9 according to section 2.2. Adjust ODL1 according to section 2.4.
- Measure the diameter of the actinic pump beam at M24 and M32 using grid paper. If the diameters are significantly different between the two positions, adjust the position of L9 parallel with the beam using a micrometer on the base plate of L9 until the diameters become almost identical.
- Remove L10 and M32. Adjust M30 and M31 according to section 2.3.
- Place P2 at the position of M32. Place a business card behind P2 as a screen.
- Set P2 at the angle that allows the pulse to be polarized at 35.3° with respect to the vertical axis to pass through P2. Rotate HWP3 until the beam spot on the screen fully disappears. Conduct this protocol for eliminating the effect of molecular reorientation on time-resolved measurements.
- Remove P2. Place M32 and reflect the beam toward the flow cell (FC). Place L10 according to section 2.2.
- Flow cell startup (Figure 1E)
- Attach a 2 mm quartz flow cell to the mount. Connect each end of the flow cell to a polyfluoroacetate (PFA) tube (length = ~500 mm; outer diameter = 1/8 inch) with an elastomer tube (length = ~10 mm).
- Insert the tube from the bottom of the flow cell to a reservoir filled with a sample solution. Attach the tube from the top of the flow cell to the inlet of the magnet gear pump.
- Attach a PFA tube (length = ~500 mm; outer diameter = 1/8 inch) to the outlet of the magnet gear pump and insert the other end to the reservoir.
- Place the flow cell mount at the focus of the probe beam.
- Turn on the magnetic gear pump. Adjust the flow rate to ~20 mL/min using the voltage control of the pump in order to replace the sample in the illuminated volume before every actinic pump pulse reaches FC.
3. Software operation
- Detector setup
- Open the Detector pane. Click the Initialize button. Wait until the Detector Initialized indicator is lit.
- Enter 40 in the Exposure Time (ms) box.
- Select IGA Lo Gain and IGA 280 kHz from the A/D gain and A/D rate drop-down menus, respectively. IGA and A/D stand for InGaAs and the analog-to-digital converter, respectively.
- Click the Set button below the Detector Set Up indicator. Confirm that the indicator light is on.
- Set the Trigger switch to External from the Trigger Event drop-down menu. Select Each – For Each Acq and TTL Rising Edge from the Trigger Edge drop-down menu. TTL stands for transistor-transistor logic.
- Click the Set button below the Trigger Set indicator. Confirm that the indicator light is on.
- Click the Read button at the bottom of the pane. Confirm that the Detector Temperature (K) box displays a value below 170 K. If not, wait until the temperature decreases below 170 K.
- Spectrograph setup
- Open the Spectrograph pane. Click the Initialize button. Wait until the Spectrograph Initialized indicator light is on.
- Select 1. Grooves 300 g/mm, Blaze Wavelength 2000 nm from the Grating drop-down menu. Click the Set button on the right-hand side of the Grating drop-down menu.
- Enter the center wavelength of the spectrograph in the Move To box and click the Go button. The center wavelength is typically located between 1,380 and 1,430 nm when the spectrograph covers the fingerprint region of the stimulated Raman spectrum.
- Enter an entrance slit width in the Set Entrance box and click the Set button on the right-hand side of the box. The entrance slit width is typically set at 0.3 mm.
- Stage position control
- Open the Visualizar pane. Enter a value of the ODL1 position in micrometers in the SK Stage Position (μm) box. The box accepts values from 0 to 200,000 (μm). Click the Go button on the right-hand side of the box.
- Enter a value of the ODL2 position in 0.1 μm in the FA Stage Position (1/10 μm) box. The box accepts values from -250,000 to 250,000 (x 1/10 μm). Click the Go button on the right-hand side of the box.
- Single measurement
- Enter the number of accumulations for a single measurement of a spectrum in the Accumulation box. The box accepts values from 1 to 999.
- Close the entrance of the spectrograph by pushing the diaphragm bar to the right as far as it can move. Click the Store Dark button. Open the entrance of the spectrograph by pulling the diaphragm bar to the left as far as it can move.
- Check the Average box to preview only an averaged result.
- Select Acquire Light Spectrum and Check Transient Absorption from the Operation Mode drop-down list for measuring probe intensities and measuring stimulated Raman or transient absorption spectra, respectively.
- Click the Acquire button.
- To automatically repeat measurements, check the Continuous box and click the Acquire button. Uncheck the Continuous box to stop the continuous measurements.
- Open the file dialog by clicking the folder icon. Double-click a folder for saving data. Enter a file name with the extension ".txt" and click OK. Click the Save button.
- Time-resolved measurement
- Open the Experiment pane. Enter a name within 20 characters that briefly describes an experiment (e.g., names of samples, conditions) in the Experiment Name box.
- Open the file dialog by clicking the folder icon. Double-click a folder for saving data and click OK.
- Enter the number of translational stage scans in the Number of Scans box.
- Select the translational stage scanned in the experiment in the Stage for Scan drop-down menu.
- Enter a stage position where the scan starts in the De box of Range A. The unit and range of the acceptable values depend on the stage (see section 3.3).
- Enter an interval between two successive stage positions in the Step box of Range A. The interval of 1 μm in the stage position corresponds to the interval of 6.7 fs in the time delay between the actinic (or Raman) pump and probe pulses.
- Enter the number of stage positions in a scan in the Points box of Range A.
- If more than one interval is required in a single scan, check the Range B box and repeat steps 3.5.5–3.5.7 for Range B. Three intervals can be set using Range A, B, and C.
- Start the scans by clicking the Run button. The Experiment Running indicator light will turn on. Wait until the indicator light turns off.
4. Optimization of probe spectrum
- Place beam dumps in the paths of the actinic and Raman pump beams. Set P1 at the angle that allows the vertically polarized pulse to pass through P1.
- Set the number of accumulations to be 10 according to step 3.4.1. Store the dark signal according to steps 3.4.2. Select Acquire Light Spectrum according to step 3.4.4.
- Run continuous measurements according to step 3.4.6 for previewing data. Maximize detector counts on the display by gradually rotating HWP1.
- Gradually increase the intensity of the incident pulse by rotating VND1 until the maximal and minimal detector counts reach around 30,000 and 4,000, respectively. If a large oscillatory pattern starts to be observed, rotate VND1 in the opposite direction until the pattern disappears.
- Stop the continuous measurements according to step 3.4.6.
5. Measurement of stationary stimulated Raman spectra
- Spatial overlap of Raman pump and probe pulses
- Remove the beam dump in the Raman pump beam path. Place the optical chopper (OC) in the Raman pump beam path.
- Place a near-IR sensor card at the sample position. Adjust the direction of the Raman pump beam by adjusting M21 according to section 2.1 until the spots of the Raman pump and probe beams fully overlap with each other. Remove the sensor card.
- Temporal overlap of Raman pump and probe pulses
- Place an InGaAs PIN photodiode at the sample position where the Raman pump and probe beams spatially overlap with each other as a result of section 5.1.
- Connect the signal output of the photodiode to a 500 MHz, 5 GS/s digital oscilloscope in order to monitor when the Raman pump and probe pulses arrive at the sample position.
- Set the horizontal scale of the oscilloscope to be 1 ns/div.
- Read the peak time of the signal intensity for the Raman pump and probe pulses blocking the other pulse.
- If a difference in the peak time is observed for the two pulses, adjust the position of ODL2 according to section 3.3 until the difference becomes smaller than 200 ps.
- Adjustment of optical chopper rotational phase
- Add 40 mL of cyclohexane to the reservoir. Start flowing cyclohexane according to step 2.9.5.
- Set the center wavelength of the spectrograph to be 1,190 nm according to step 3.2.3 to observe Rayleigh scattering of the Raman pump pulse.
- Set the number of accumulations to 10 according to step 3.4.1. Store the dark signal according to step 3.4.2.
- Select Check Transient Absorption according to step 3.4.4.
- Run continuous measurements according to step 3.4.6.
- Maximize the amplitude of the apparent transient absorption signal with the negative sign at the Raman pump wavelength, which originates from the presence and absence of the scattered Raman pump pulse due to chopping, by adjusting the rotational phase of OC from -180°−170° on the front panel of the controller.
- Stop the continuous measurements according to step 3.4.6.
- Signal maximization
- Set the center wavelength of the spectrograph to be 1,410 nm according to step 3.2.3 for observing stimulated Raman spectra.
- Run continuous measurements according to step 3.4.6 and check if stimulated Raman bands of cyclohexane are observed in the display. The strongest band of cyclohexane appears at the 55th–58th pixels when the center wavelength is set at 1,410 nm.
- If the stimulated Raman bands are not observed, try to change the position of ODL2 by ±15,000 μm at 150 μm intervals according to section 3.3 and see if the stimulated Raman bands are observed.
- If the stimulated Raman bands are not observed after step 5.4.3 is conducted, retry step 5.1.2 to obtain the spatial overlap between the Raman pump and probe beams and conduct step 5.4.2 again.
- Once the stimulated Raman bands are detected, maximize the band intensities in the display by iteratively readjusting M21, the rotational phase of OC, and the position of ODL2.
- Stop the continuous measurements according to step 3.4.6.
- Measurement
- Set the number of accumulations to be 500 according to step 3.4.1. Store the dark signal according to step 3.4.2.
- Run a single measurement according to step 3.4.5. Save the spectrum according to step 3.4.7. Repeat the measurement at least 4x.
- Remove the FC inlet tube from the reservoir and wait until the flow is interrupted by the air. Minimize the voltage of the magnetic gear pump.
- Replace the content of the reservoir with the one filled with 100 mL of fresh acetone.
- Set the inlet and outlet tubes into the reservoir and empty flask, respectively. Start the magnetic gear pump according to step 2.9.5 and let toluene flow through the FC.
- Wait until the flow is interrupted by the air. Minimize the voltage of the magnetic gear pump.
- Repeat steps 5.5.4–5.5.6 at least 2x.
- Add 40 mL of acetone to the reservoir. Start flowing acetone according to step 2.9.5.
- Record the stimulated Raman spectrum of acetone according to step 5.5.2.
- Remove acetone from the FC according to step 5.5.3.
- Repeat steps 5.5.4–5.5.10 using toluene instead of acetone.
6. Measurement of time-resolved absorption spectra
- Empty the reservoir and add 25 mL of toluene solution of β-carotene with a concentration of 1 x 10-4 mol dm-3. Start flowing the sample solution according to step 2.9.5.
- Place the OC in the actinic pump beam path.
- Move the beam dump from the path of the actinic pump beam to that of the Raman pump beam.
- Spatially overlap the actinic pump and probe beams at the sample position according to step 5.1.2 using a business card instead of the near-IR sensor card.
- Temporally overlap the two beams at the sample position according to section 5.2 using a Si PIN photodiode instead of the InGaAs PIN photodiode.
- Set the number of accumulations to be 10 according to step 3.4.1. Store the dark signal according to step 3.4.2.
- Select Check Transient Absorption according to step 3.4.4.
- Run continuous measurements according to step 3.4.6 and check if the transient absorption of β-carotene is observed in the display. The absorption band appears with a shape decreasing monotonically towards longer wavelengths (the second lowest excited singlet state, S2) or with two maxima at around the 0th and 511th pixels (the lowest excited singlet state, S1).
- If the transient absorption is not observed, try to change the position of ODL1 by ±15,000 μm at 150 μm intervals according to section 3.3.
- If no absorption band is observed after step 6.9 is conducted, retry step 6.4 to obtain the spatial overlap between the actinic pump and probe beams.
- Maximize the absorption intensity by readjusting M32 once the transient absorption band is detected.
- Stop the continuous measurements according to step 3.4.6.
- Decrease the position of ODL1 according to section 3.3 until the transient absorption fully disappears.
7. Measurement of time-resolved stimulated Raman spectra
- Place the OC in the Raman pump beam path. Remove the beam dump from the Raman pump beam path.
- Set the number of accumulations to 200 according to step 3.4.1. Store the dark signal according to step 3.4.2.
- Run a time-resolved experiment according to section 3.5. In step 3.5.4, select SK stage. Set the Start value of Range A to be smaller by around 50 μm than the position where the transient absorption signal disappeared in step 6.13.
8. Raman shift calibration
- Calculate the average of the four stimulated Raman spectra for cyclohexane, acetone, and toluene recorded in section 5 using data analysis software of your choice.
- Plot the averaged stimulated Raman spectra of the solvents against the pixel number of the InGaAs array detector.
- Estimate the peak positions of the stimulated Raman bands of the solvents by least-squares fitting analysis with the Lorentzian functions. If the Lorentzian function is not available, use a polynomial function instead.
- Plot the peak wavenumbers of the Raman bands of the solvents in a reference book (e.g., Hamaguchi and Iwata34) against the estimated peak positions in the pixel number.
- Obtain a calibration function between the Raman shift and the pixel number by the least-squares fitting analysis with a second- or third-degree polynomial function.
Representative Results
Femtosecond time-resolved near-IR stimulated Raman spectroscopy was applied to β-carotene in toluene solution. The concentration of the sample was 1 x 10-4 mol dm-3. The sample was photoexcited by the actinic pump pulse at 480 nm with a pulse energy of 1 μJ. Time-resolved stimulated Raman spectra of β-carotene in toluene are shown in Figure 2A. The raw spectra contained strong Raman bands of the solvent toluene and a weak Raman band of β-carotene in the ground state as well as Raman bands of photoexcited β-carotene. They were subtracted using the stimulated Raman spectrum of the same solution at 1 ps before photoexcitation. The spectra after the subtraction (Figure 2B) showed distorted baselines that are caused by absorption of photoexcited β-carotene and/or other nonlinear optical processes. The baselines became flat after they were corrected with polynomial functions (Figure 2C).
The time-resolved stimulated Raman spectra of β-carotene showed two strong bands in the 1,400–1,800 cm-1 region (Figure 2C). A broad stimulated Raman band at 0 ps was assigned to the in-phase C=C stretch vibration of S2 β-carotene. Its peak position was estimated to be 1,556 cm-1. The in-phase C=C stretch band of S1 β-carotene appeared as the S2 C=C stretch band decayed. The peak position of the S1 C=C stretch band was upshifted by 8 cm-1 from 0.12 to 5 ps (Figure 2D). The time constant of the upshift was estimated to be 0.9 ps. The upshift originates from vibrational energy redistribution in S1 β-carotene2,3.
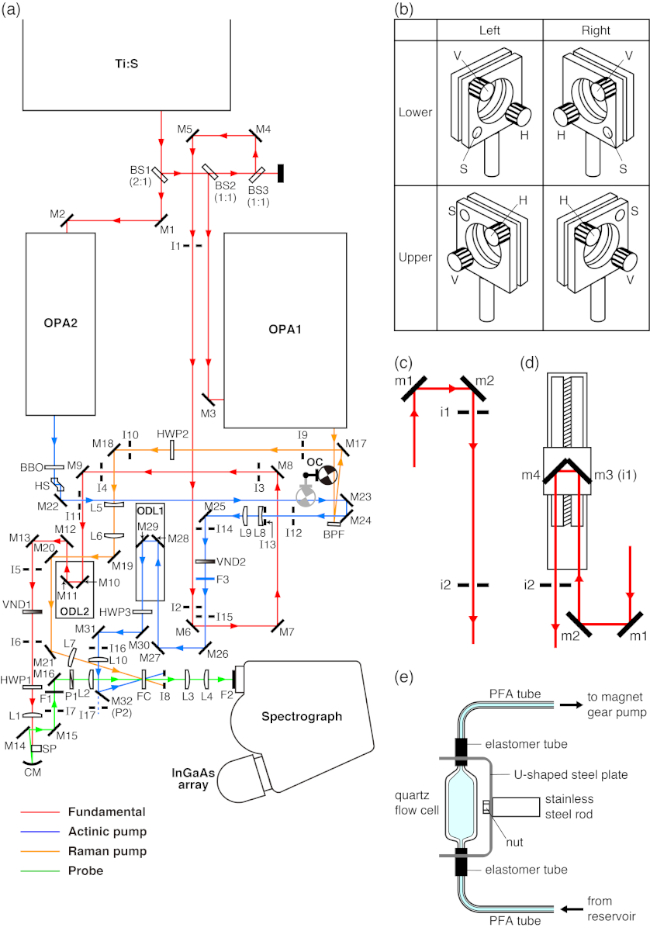
Figure 1: Instrument diagrams. (A) The block diagram of a femtosecond time-resolved near-IR stimulated Raman spectrometer. Ti:S = Mode-locked Ti:sapphire laser system; BS = Beamsplitter; OPA = Optical parametric amplifier; BBO = β-Barium borate crystal; OC = Optical chopper; ODL = Optical delay line; BPF = Volume-grating reflective bandpass filter; SP = Sapphire plate; FC = Flow cell; M = Mirror; CM = Concave mirror; L = Lens; I = Iris; P = Polarizer; HWP = Half-wave plate; F = Color glass filter; VND = Variable optical density filter. The figure is adapted from Takaya11 with permission from the PCCP Owner Societies. (B) Four configurations of a mirror mount. V, H, and S represent the vertical adjustment knob, horizontal adjustment knob, and support, respectively. See section 2.1 for details. (C) A schematic diagram of laser beam alignment. m = Mirror; i = Iris. See section 2.3 for details. (D) A schematic diagram of optical delay line alignment. m = Mirror; i = Iris. See section 2.4 for details. (E) Structure of a flow cell mount. See section 2.9 for details. Please click here to view a larger version of this figure.
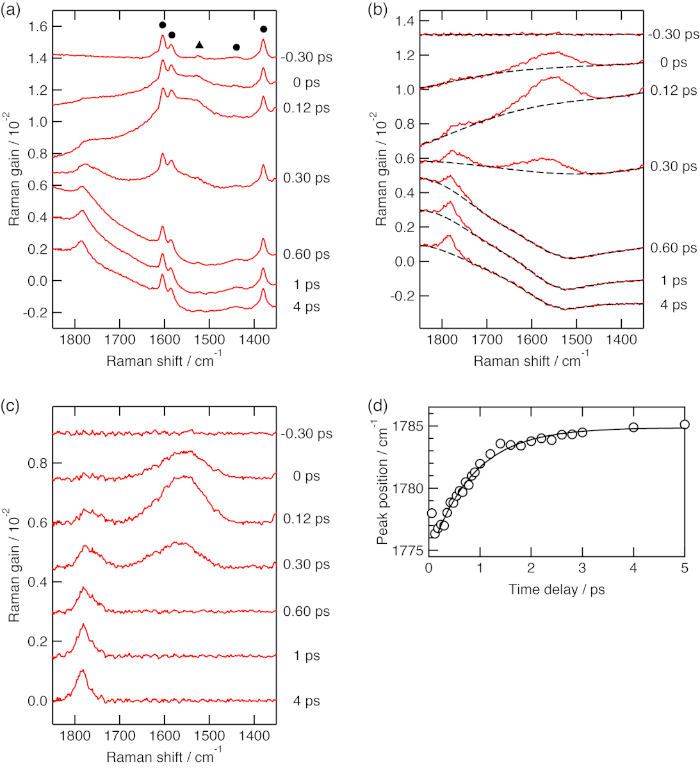
Figure 2: Femtosecond time-resolved near-IR stimulated Raman spectra. (A) Femtosecond time-resolved near-IR stimulated Raman spectra of β-carotene in toluene with the actinic pump wavelength at 480 nm. Raman bands of toluene and β-carotene in the ground state are denoted with circles and a triangle, respectively. (B) Femtosecond time-resolved near-IR stimulated Raman spectra of β-carotene in toluene after the Raman bands of toluene and β-carotene in the ground state are subtracted. The baselines of the spectra were fitted with polynomial functions (broken traces). (C) Femtosecond time-resolved near-IR stimulated Raman spectra of β-carotene in toluene after the baseline correction. (D) The peak positions of the in-phase C=C stretch band in the S1 state plotted against the time delay. The C=C stretch bands were fitted with a Gaussian function for estimating their peak positions. The best fitted curve for the shift of the S1 C=C stretch band (solid trace) was obtained by the least-squares fitting analysis with an exponential function. Please click here to view a larger version of this figure.
Discussion
Crucial factors in femtosecond time-resolved near-IR multiplex stimulated Raman measurement
To obtain time-resolved near-IR stimulated Raman spectra with a high signal-to-noise ratio, the probe spectrum should ideally have uniform intensity in the whole wavelength range. White-light continuum generation (section 2.5) is, therefore, one of the most crucial parts of time-resolved near-IR stimulated Raman experiments. In general, the probe spectrum becomes broad and flat as the intensity of the incident beam increases. A high beam intensity, however, easily produces undesirable nonlinear optical effects other than the white light continuum generation. In a worst-case scenario, the nonlinear effects provide the probe spectrum with a large intensity fluctuation and an oscillatory pattern that significantly lowers the signal-to-noise ratio of stimulated Raman spectra. Figure 2C shows how the oscillatory pattern affects the spectra. It shows oscillatory patterns from -0.30 to 4 ps, but the patterns appear only weakly, with a peak-to-peak amplitude of 1 x 10-4, as white light generation is carefully optimized. Another undesirable effect on the probe spectrum can be provided by water vapor in the air2,11. The effect of water vapor might be avoided if part of the spectrometer, including the white light generation optics, sample, and spectrograph, is set in a chamber filled with dry nitrogen.
Accuracy of Raman shift calibration
As described in section 8, we calibrate the Raman shift axis by the least-squares fitting analysis of the peak positions of the solvent bands in Raman shift against those in the pixel number of the detector with a polynomial function. We think this protocol works well as long as the Raman pump wavelength cannot be determined with high accuracy. It is the case for our spectrometer because each pixel of our detector covers as large as 3.5 cm-1 at around the wavenumber of the Raman pump pulse. However, the solvents must be chosen so that all the transient stimulated Raman bands of the sample appear between the highest and lowest wavenumbers of the solvent bands (section 8). The Raman shift calibration curve loses its accuracy beyond the range of the solvent bands. In Figure 2, a Raman band of S1 β-carotene in toluene, at 1,785 cm-1, appears beyond the highest wavenumber of the solvent bands, 1,710 cm-1. We have confirmed that the peak position agrees well with that in benzene determined by picosecond time-resolved spontaneous Raman spectroscopy35,36.
Effectiveness and perspective of femtosecond time-resolved near-IR multiplex stimulated Raman spectrometer
It has been demonstrated that the femtosecond time-resolved near-IR multiplex stimulated Raman spectrometer can observe stimulated Raman spectra, which provides information almost equivalent to spontaneous Raman spectra of short-lived species with near-IR transitions. Small differences in the peak position of a band can be detected with the spectrometer because of its sufficiently high sensitivity. The spectrometer will be applicable to a wide variety of π-conjugate systems from simple aromatic molecules to photoconductive polymers. Stationary near-IR multiplex stimulated Raman spectroscopy is also a powerful tool for observing molecular vibrations without fluorescence interference from the sample, because the energy of near-IR photons is generally much lower than the electronic transition energy of molecules from the lowest excited singlet state to the ground state. The spectrometer will be applicable to in vivo observation of the structural dynamics in biological systems.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP24750023, JP24350012, MEXT KAKENHI Grant Numbers JP26104534, JP16H00850, JP26102541, JP16H00782, and MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2015–2019.
Materials
| 1-Axis Translational Stage | OptSigma | TSD-401S | Products equivalent to this are used as well; for M22, L9, and CM in Figure 1A |
| 20-cm Optical Delay Line | OptSigma | SGSP26-200 | ODL1 in Figure 1A |
| 3-Axis Translational Stage | OptSigma | TSD-405SL | For L8 in Figure 1A |
| 3-Axis Translational Stage | Suruga Seiki | B72-40C | For FC in Figure 1A |
| 5-cm Optical Delay Line | PMT | HRS-0050 | ODL2 in Figure 1A |
| Al Concave Mirror | Thorlabs | CM254-050-G01 | Focal length: 50 mm; CM in Figure 1A |
| Base Plate | Suruga Seiki | A21-6 | Products equivalent to this are used as well; for M1-M32, BS1-BS3, L1-L10, I1-I17, P1-P2, HWP1-3, F1-F3, VND1-VND2, OC, BPF, HS, BBO, SP, CM, and FC in Figure 1A |
| BBO Crystal | EKSMA Optics | – | Type 1, θ = 23.2 deg; BBO in Figure 1A |
| BK7 Plano-Concave Lens | OptSigma | SLB-25.4-50NIR2 | Focal length: 50 mm; IR anti-reflection coating; L6 in Figure 1A |
| BK7 Plano-Convex Lens | OptSigma | SLB-25.4-150PIR2 | Focal length: 150 mm; IR anti-reflection coating; L2, L3, L5 in Figure 1A |
| BK7 Plano-Convex Lens | OptSigma | SLB-25.4-100PIR2 | Focal length: 100 mm; IR anti-reflection coating; L4 in Figure 1A |
| BK7 Plano-Convex Lens | OptSigma | SLB-25.4-200PIR2 | Focal length: 200 mm; IR anti-reflection coating; L7 in Figure 1A |
| Broadband Dielectric Mirror | OptSigma | TFMS-25.4C05-2/7 | M22-M25, M28, M29 in Figure 1A |
| Broadband Dielectric Mirror | Precision Photonics (Advanced Thin Films) | – | M26, M27, M30-M32 in Figure 1A |
| Broadband Half-Wave Plate | CryLight | – | HWP3 in Figure 1A |
| Color Glass Filter | HOYA | IR85 | F1 in Figure 1A |
| Color Glass Filter | HOYA | RM100 | F2 in Figure 1A |
| Color Glass Filter | Schott | BG39 | F3 in Figure 1A |
| Computer | Dell | Vostro 200 Mini Tower | OS: Windows XP |
| Cyclohexane | Kanto Kagaku | 07547-1B | HPLC grade |
| Data Analysis Software | Wavemetrics | Igor Pro 8 | |
| Dielectric Beamsplitter | LAYERTEC | – | Reflection : Transmission = 2 : 1; BS1 in Figure 1A |
| Dielectric Beamsplitter | LAYERTEC | – | Reflection : Transmission = 1 : 1; BS2, BS3 in Figure 1A |
| Dielectric Mirror | Precision Photonics (Advanced Thin Films) |
– | M1-M8 in Figure 1A |
| Digital Oscilloscope | Tektronix | TDS3054B | 500 MHz, 5 GS/s |
| Elastomer Tube | – | – | Figure 1E |
| Femtosecond Ti:sapphire Oscillator | Coherent | Vitesse 800-2 | Wavelength: 800 nm, pulse duration: 100 fs, average power: 280 mW, repetition rate: 80 MHz; included in Ti:S in Figure 1A |
| Femtosecond Ti:sapphire Regenerative Amplifier | Coherent | Legend-Elite-F-HE | Wavelength: 800 nm, pulse duration: 100 fs, pulse energy: 3.5 mJ, repetition rate: 1 kHz; included in Ti:S in Figure 1A |
| Film Polarizer | OptSigma | SPFN-30C-26 | P1 in Figure 1A |
| Glan-Taylor Prism | OptSigma | GYPB-10-10SN-3/7 | P2 in Figure 1A |
| Gold Mirror | OptSigma | TFG-25C05-10 | M9-M21 in Figure 1A |
| Half-Wave Plate | OptSigma | WPQ-7800-2M | HWP1 in Figure 1A |
| Harmonic Separator | Coherent | TOPAS-C HRs 410-540 nm | HS in Figure 1A |
| InGaAs Array Detector | Horiba | Symphony-IGA-512X1-50-1700-1LS | 512 ch, Liquid nitrogen cooled |
| InGaAs PIN Photodiode | Hamamatsu Photonics | G10899-01K | |
| IR Half-Wave Plate | OptiSource | – | HWP2 in Figure 1A |
| Iris | Suruga Seiki | F74-3N | Products equivalent to this are used as well; I1-I17 in Figure 1A |
| Lens Holder | OptSigma | LHF-25.4S | Products equivalent to this are used as well; for L1-L10 in Figure 1A |
| Magnetic Gear Pump | Micropump | 184-415 | |
| Mirror Mount | Siskiyou | IM100.C2M6R | Products equivalent to this are used as well; for M1-M32, BS1-BS3, BBO, CM in Figure 1A |
| near-IR phosphor card | Thorlabs | VRC2 | |
| Nut | – | – | Figure 1E, M4; purchased from a DIY store |
| Optical Chopper | New Focus | 3501 | OC in Figure 1A |
| Optical Parametric Amplifier | Coherent | OPerA-F | OPA1 in Figure 1A |
| Optical Parametric Amplifier | Coherent | TOPAS-C | OPA2 in Figure 1A |
| Polarizer Holder | OptSigma | PH-30-ARS | Products equivalent to this are used as well; for P1-P2 and HWP1-3 In Figure 1A |
| Polyfluoroacetate Tube | – | – | Figure 1E |
| Post Holder | OptSigma | BRS-12-80 | Products equivalent to this are used as well; for M1-M32, BS1-BS3, L1-L10, I1-I17, P1-P2, HWP1-3, F1-F3, VND1-VND2, OC, BPF, HS, BBO, SP, CM, and FC in Figure 1A |
| Quartz Flow Cell | Tosoh Quartz | T-70-UV-2 | FC in Figure 1A |
| Quartz Plano-Concave Lens | OptSigma | SLSQ-25-50N | Focal length: 50 mm; L8 in Figure 1A |
| Quartz Plano-Convex Lens | OptSigma | SLSQ-25-100P | Focal length: 100 mm; L1, L9 in Figure 1A |
| Quartz Plano-Convex Lens | OptSigma | SLSQ-25-220P | Focal length: 220 mm; L10 in Figure 1A |
| Sapphire Plate | Pier Optics | – | 3 mm thick; SP in Figure 1A |
| Si PIN Photodiode | Hamamatsu Photonics | S3883 | |
| Single Spectrograph | Horiba Jobin Yvon | iHR320 | Focal length: 32 cm |
| Stainless Steel Rod | Suruga Seiki | A41-100 | Products equivalent to this are used as well; for M1-M32, BS1-BS3, L1-L10, I1-I17, P1-P2, HWP1-3, F1-F3, VND1-VND2, OC, BPF, HS, BBO, SP, CM, and FC in Figure 1A |
| Stainless Steel Rod | Newport | J-SP-2 | Figure 1E |
| Toluene | Kanto Kagaku | 40180-1B | HPLC grade |
| U-Shaped Steel Plate | – | – | Figure 1E; purchased from a DIY store |
| Variable Neutral Density Filter (with a holder) | OptSigma | NDHN-100 | VND1 in Figure 1A |
| Variable Neutral Density Filter (with a holder) | OptSigma | NDHN-U100 | VND2 in Figure 1A |
| Visual Programming Language | National Instruments | LabVIEW 2009 | The control software in this study is programmed in LabVIEW 2009 |
| Volume-Grating Bandpass Filter | OptiGrate | BPF-1190 | BPF in Figure 1A |
| β-Carotene | Wako Pure Chemical Industries | 035-05531 |
Referências
- Polívka, T., Herek, J. L., Zigmantas, D., Åkerlund, H. -. E., Sundström, V. Direct Observation of the (Forbidden) S1 State in Carotenoids. Proceedings of the National Academy of Sciences of the United States of America. 96 (9), 4914-4917 (1999).
- Takaya, T., Iwata, K. Relaxation Mechanism of β-Carotene from S2 (1Bu+) State to S1 (2Ag-) State: Femtosecond Time-Resolved Near-IR Absorption and Stimulated Resonance Raman Studies in 900-1550 nm Region. Journal of Physical Chemistry A. 118 (23), 4071-4078 (2014).
- Takaya, T., Anan, M., Iwata, K. Vibrational Relaxation Dynamics of β-Carotene and Its Derivatives with Substituents on Terminal Rings in Electronically Excited States as Studied by Femtosecond Time-Resolved Stimulated Raman Spectroscopy in the Near-IR Region. Physical Chemistry Chemical Physics. 20 (5), 3320-3327 (2017).
- Guo, J., Ohkita, H., Benten, H., Ito, S. Near-IR Femtosecond Transient Absorption Spectroscopy of Ultrafast Polaron and Triplet Exciton Formation in Polythiophene Films with Different Regioregularities. Journal of the American Chemical Society. 131 (46), (2009).
- Hwang, I. -. W., et al. Carrier Generation and Transport in Bulk Heterojunction Films Processed with 1,8-Octanedithiol as a Processing Additive. Journal of Applied Physics. 104 (3), 033706 (2008).
- Yonezawa, K., Kamioka, H., Yasuda, T., Han, L., Moritomo, Y. Fast Carrier Formation from Acceptor Exciton in Low-Gap Organic Photovoltaic. Applied Physics Express. 5 (4), 042302 (2012).
- Takaya, T., Enokida, I., Furukawa, Y., Iwata, K. Direct Observation of Structure and Dynamics of Photogenerated Charge Carriers in Poly(3-hexylthiophene) Films by Femtosecond Time-Resolved Near-IR Inverse Raman Spectroscopy. Molecules. 24 (3), 431 (2019).
- Clafton, S. N., Huang, D. M., Massey, W. R., Kee, T. W. Femtosecond Dynamics of Excitons and Hole-Polarons in Composite P3HT/PCBM Nanoparticles. Journal of Physical Chemistry B. 117 (16), 4626-4633 (2013).
- Cook, S., Furube, A., Katoh, R. Analysis of the Excited States of Regioregular Polythiophene P3HT. Energy & Environmental Science. 1 (2), 294-299 (2008).
- Okino, S., Takaya, T., Iwata, K. Femtosecond Time-Resolved Near-Infrared Spectroscopy of Oligothiophenes and Polythiophene: Energy Location and Effective Conjugation Length of Their Low-Lying Excited States. Chemistry Letters. 44 (8), 1059-1061 (2015).
- Takaya, T., Iwata, K. Development of a Femtosecond Time-Resolved Near-IR Multiplex Stimulated Raman Spectrometer in Resonance with Transitions in the 900-1550 nm Region. Analyst. 141 (14), 4283-4292 (2016).
- Jas, G. S., Wan, C., Johnson, C. K. Picosecond Time-Resolved Fourier Transform Raman Spectroscopy of 9,10-Diphenylanthracene in the Excited Singlet State. Applied Spectroscopy. 49 (5), 645-649 (1995).
- Jas, G. S., Wan, C., Kuczera, K., Johnson, C. K. Picosecond Time-Resolved Fourier-Transform Raman Spectroscopy and Normal-Mode Analysis of the Ground State and Singlet Excited State of Anthracene. Journal of Physical Chemistry. 100 (29), 11857-11862 (1996).
- Sakamoto, A., Okamoto, H., Tasumi, M. Observation of Picosecond Transient Raman Spectra by Asynchronous Fourier Transform Raman Spectroscopy. Applied Spectroscopy. 52 (1), 76-81 (1998).
- Sakamoto, A., Matsuno, S., Tasumi, M. Construction of Picosecond Time-Resolved Raman Spectrometers with Near-Infrared Excitation. Journal of Raman Spectroscopy. 37 (1-3), 429-435 (2006).
- Sakamoto, A., Matsuno, S., Tasumi, M. Picosecond Near-Infrared Excited Transient Raman Spectra of β-Carotene in the Excited S2 State: Solvent Effects on the in-Phase C=C Stretching Band and Vibronic Coupling. Journal of Molecular Structure. 976 (1-3), 310-313 (2010).
- Yoshizawa, M., Kurosawa, M. Femtosecond Time-Resolved Raman Spectroscopy Using Stimulated Raman Scattering. Physical Review A. 61 (1), 013808 (2000).
- Yoshizawa, M., Kubo, M., Kurosawa, M. Ultrafast Photoisomerization in DCM Dye Observed by New Femtosecond Raman Spectroscopy. Journal of Luminescence. 87-89, 739-741 (2000).
- Yoshizawa, M., Aoki, H., Hashimoto, H. Vibrational Relaxation of the 2Ag– Excited State in All-Trans-β-Carotene Obtained by Femtosecond Time-Resolved Raman Spectroscopy. Physical Review B. 63 (18), 180301 (2001).
- McCamant, D. W., Kukura, P., Mathies, R. A. Femtosecond Broadband Stimulated Raman: A New Approach for High-Performance Vibrational Spectroscopy. Applied Spectroscopy. 57 (11), 1317-1323 (2003).
- McCamant, D. W., Kukura, P., Yoon, S., Mathies, R. A. Femtosecond Broadband Stimulated Raman Spectroscopy: Apparatus and Methods. Review of Scientific Instruments. 75 (11), 4971-4980 (2004).
- Kukura, P., McCamant, D. W., Mathies, R. A. Femtosecond Stimulated Raman Spectroscopy. Annual Review of Physical Chemistry. 58, 461-488 (2007).
- Laimgruber, S., Schachenmayr, H., Schmidt, B., Zinth, W., Gilch, P. A Femtosecond Stimulated Raman Spectrograph for the Near Ultraviolet. Applied Physics B. 85 (4), 557-564 (2006).
- Umapathy, S., Lakshmanna, A., Mallick, B. Ultrafast Raman Loss Spectroscopy. Journal of Raman Spectroscopy. 40 (3), 235-237 (2009).
- Mallick, B., Lakshmanna, A., Umapathy, S. Ultrafast Raman Loss Spectroscopy (URLS): Instrumentation and Principle. Journal of Raman Spectroscopy. 42 (10), 1883-1890 (2011).
- Kloz, M., van Grondelle, R., Kennis, J. T. M. Wavelength-Modulated Femtosecond Stimulated Raman Spectroscopy-Approach towards Automatic Data Processing. Physical Chemistry Chemical Physics. 13 (40), 18123-18133 (2011).
- Kloz, M., Weißenborn, J., Polívka, T., Frank, H. A., Kennis, J. T. M. Spectral Watermarking in Femtosecond Stimulated Raman Spectroscopy: Resolving the Nature of the Carotenoid S* State. Physical Chemistry Chemical Physics. 18 (21), 14619-14628 (2016).
- Kuramochi, H., Takeuchi, S., Tahara, T. Ultrafast Structural Evolution of Photoactive Yellow Protein Chromophore Revealed by Ultraviolet Resonance Femtosecond Stimulated Raman Spectroscopy. Journal of Physical Chemistry Letters. 3 (15), 2025-2029 (2012).
- Wang, S., et al. Dynamic High Pressure Induced Strong and Weak Hydrogen Bonds Enhanced by Pre-Resonance Stimulated Raman Scattering in Liquid Water. Optics Express. 25 (25), 31670-31677 (2017).
- Ashner, M. N., Tisdale, W. A. High Repetition-Rate Femtosecond Stimulated Raman Spectroscopy with Fast Acquisition. Optics Express. 26 (14), 18331-18340 (2018).
- Quincy, T. J., Barclay, M. S., Caricato, M., Elles, C. G. Probing Dynamics in Higher-Lying Electronic States with Resonance-Enhanced Femtosecond Stimulated Raman Spectroscopy. Journal of Physical Chemistry A. 122 (42), 8308-8319 (2018).
- Taylor, M. A., et al. Delayed Vibrational Modulation of the Solvated GFP Chromophore into a Conical Intersection. Physical Chemistry Chemical Physics. 21 (19), 9728-9739 (2019).
- Cassabaum, A. A., Silva, W. R., Rich, C. C., Frontiera, R. R. Orientation and Polarization Dependence of Ground- and Excited-State FSRS in Crystalline Betaine-30. Journal of Physical Chemistry C. 123 (20), 12563-12572 (2019).
- Hamaguchi, H., Iwata, K. . Raman Spectroscopy (The Spectroscopical Society of Japan, Spectroscopy Series 1). , (2015).
- Hashimoto, H., Koyama, Y. The C=C Stretching Raman Lines of β-Carotene Isomers in the S1 State as Detected by Pump-Probe Resonance Raman Spectroscopy. Chemical Physics Letters. 154 (4), 321-325 (1989).
- Noguchi, T., Hayashi, H., Tasumi, M., Atkinson, G. H. Solvent Effects on the ag C=C Stretching Mode in the 21Ag- Excited State of β-Carotene and Two Derivatives: Picosecond Time-Resolved Resonance Raman Spectroscopy. Journal of Physical Chemistry. 95 (8), 3167-3172 (1991).
