1. Finding the minimum energy structure of isolated glycine and water
NOTE: The goal here is two-fold: (i) to obtain minimum energy structures of isolated water and glycine molecules for use in the genetic algorithm configurational sampling, (ii) and to compute the thermodynamic corrections to the gas phase energies of these molecules for use in the calculation of atmospheric concentrations.
- On your local computer, open a new session of Avogadro.
- Click Build > Insert > Peptide and select Gly from the Insert Peptide window to generate a glycine monomer in the visualization window.
- Click Extensions > Gaussian and edit the first line in the text box to read '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(conver=12) opt(tight,maxcyc=300) freq'. Click Generate and save the input file as glycine.com.
- Please note that if the molecule has significant conformational flexibility, as glycine does55, it is critical to perform conformational analysis to identify the global minimum structure and other low-lying conformers. OpenBabel54 provides robust conformational search tools utilizing different algorithms and quick force fields. While conformers are allowed to relax and interconvert during GA and subsequent calculations, it is sometimes necessary to run multiple GA calculations, each starting with a different conformer.
- On your local computer, open a new session in Avogadro.
- Click Build > Insert > Fragment and search for "water" from the Insert Fragment window to get the coordinates of water.
- Click Extensions > Gaussian and edit the first line in the text box to read '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(conver=12) opt(tight,maxcyc=300) freq'. Click Generate and save the input file as water.com.
- Transfer the two .com files to the remote cluster. Once you log into the remote cluster, call Gaussian 09 in a batch submission script to start the calculation. When the calculations finish, extract the Cartesian coordinates (.xyz files) of the minimum energy structures by calling OpenBabel. For glycine, the command to execute is:
obabel -ig09 glycine.log -oxyz > glycine.xyz
These two .xyz files will be used by the GA configurational sampling in the next step.
2. Genetic-algorithm-based configurational sampling of Gly(H2O)n=1-5 clusters
NOTE: The goal here is to obtain a set of low-energy structures for Gly(H2O)n=1-5 at the inexpensive semi-empirical level of theory, using the PM746 model implemented in MOPAC47. It is imperative that the working directory has the exact organization and structure as shown in Figure 2. This is to ensure that the custom shell and Python scripts work without failures.
- Copy all necessary scripts to the remote cluster and add their location to $PATH
- Put all scripts and template files to a folder (Eg. scripts) and copy it to the remote cluster
- Make sure all the scripts are executable
- Add the location of the scripts directory to the $PATH environmental variable by entering the following commands in a terminal. The default location of the scripts is set to $HOME/JoVE-demo/scripts, however, one can define an environmental variable called $SCRIPTS_HOME pointing to the directory containing the scripts and add $SCRIPTS_HOME to one's path
- Bash shell:
export SCRIPTS_HOME=/path/to/scripts
export PATH=${SCRIPTS_HOME}:${PATH} - Tcsh/Csh shell:
setenv SCRIPTS_HOME /path/to/scripts
setenv PATH ${SCRIPTS_HOME}:${PATH}
- Bash shell:
- On the remote cluster, set up and run a GA calculation:
- Create a directory called gly-h2o-n where n is the number of water molecules.
- Create a subdirectory called GA under the gly-h2o-n directory to run genetic algorithm calculations.
- Copy the OGOLEM input files (Eg. pm7.ogo), monomers Cartesian coordinates (Eg. glycine.xyz, water.xyz) and PBS batch submission script (Eg. run.pbs) into the GA directory.
- Make the necessary changes to the OGOLEM input file and batch submission file.
- Submit the calculation. When the calculation starts, OGOLEM will create a new directory named as the prefix of the OGOLEM input file (Eg. pm7) in the GA directory and store newly generated coordinates there.
- Once the calculation is complete, compile the energies and rotational constants, and use that information to determine which are the unique low-energy structures:
- Change directory to gly-h2o-n/GA/pm7 and
- Extract the energies and compute the rotational constants of the GA-optimized clusters with the command:
getRotConsts-GA.csh N 0 99
where N is the number of atoms in the molecular cluster and '0 99' indicates that the GA pool size is 100, with indices running from 0 through 99. This will generate a file called rotConstsData_C which contains a sorted list of all the GA-optimized cluster configurations, their energies, and their rotational constants. - Execute the command:
similarityAnalysis.py pm7 rotConstsData_C
where pm7 will be used as a file-naming label, to find and save the unique GA-optimized clusters. This will generate a file called uniqueStructures-pm7.data which contains a sorted list of the unique GA-optimized configurations. This is a list of unique local minimum structures for the Gly(H2O)n cluster optimized at the PM7 level of theory, and these structures are now ready to be refined using DFT.
- Go up to the gly-h2o-n/GA directory and combine the results from multiple comparable GA runs using the combine-GA.csh script. The syntax is:
combine-GA.csh <label> <list of directories with GA runs>
In this particular case, the command:
combine-GA.csh pm7 pm7
will generate a new unique structures list named 'uniqueStructures-pm7.data' in the gly-h2o-n/GA directory.
3. Refinement using QM method with a small basis set
NOTE: The goal here is to refine the configurational sampling of the Gly(H2O)n=1-5 clusters using a better quantum-mechanical description to obtain a smaller but more accurate set of Gly(H2O)n=1-5 cluster structures. The starting structures for this step are the outputs of Step 2.
- Prepare and run the small basis set DFT calculation:
- Create a subdirectory called QM under the gly-h2o-n directory. Under the QM directory, create another subdirectory named pw91-sb.
- Copy the unique structures list (uniqueStructures-pm7.data) from the gly-h2o-n/GA directory to the QM/pw91-sb directory.
- Change directory to that gly-h2o-n/QM/pw91-sb.
- Run the small basis set DFT configurational sampling script using the command:
run-pw91-sb.csh uniqueStructures-pm7.data sb QUEUE 10
where sb is a label for this set of calculations, QUEUE is the preferred queue on the computing cluster, and 10 indicates that 10 calculations are to be grouped into one batch job. This script will automatically generate the inputs for Gaussian 09 and submit all the calculations. Enter 'test' for the 'QUEUE' to do a dry run.
- Once the submitted calculations are complete, extract and analyze the results.
- Extract the energies and compute the rotational constants of the small-basis-optimized clusters using the command:
getRotConsts-dft-sb.csh pw91 N
where pw91 indicates that the PW91 density functional was used, and N is the number of atoms in the cluster. That will create a file named rotConstsData_C. - Now identify the unique structures with the command:
similarityAnalysis.py sb rotConstsData_C
where sb is used as a file-naming label. There will now be a list of unique configurations optimized at the PW91/6-31+G* level of theory saved in the file uniqueStructures-sb.data.
- Extract the energies and compute the rotational constants of the small-basis-optimized clusters using the command:
- Go up to the gly-h2o-n/QM directory and combine the results from multiple comparable QM runs using the combine-QM.csh script. The syntax is:
combine-QM.csh <label> <list of directories with QM calcs>
In this particular case, the command:
combine-QM.csh sb pw91-sb
will generate a new unique structures list named 'uniqueStructures-sb.data' in the gly-h2o-n/QM directory.
4. Further refinement using QM method with a large basis set
NOTE: The goal here is to further refine the configurational sampling of the Gly(H2O)n=1-5 clusters using a better quantum-mechanical description. The starting structures for this step are the outputs of Step 3.
- Submit more reliable calculations using a larger basis set.
- Create a subdirectory called pw91-lb under the QM directory.
- Copy the unique structures list (uniqueStructures-sb.data) from the gly-h2o-n/QM directory to the gly-h2o-n/QM/pw91-lb directory and change to that directory.
- Run the large-basis DFT configurational sampling script with the command:
run-pw91-lb.csh uniqueStructures-sb.data lb QUEUE 10
where lb is a label for this set of calculations, QUEUE is the preferred queue on the computing cluster, and 10 indicates that 10 calculations are to be grouped into one batch job. This script will automatically generate the inputs for Gaussian 09 and submit all the calculations. Enter 'test' for the 'QUEUE' to do a dry run testing.
- Once the submitted calculations are complete, extract and analyze the data
- Compute the rotational constants of the large-basis-optimized clusters with the command:
getRotConsts-dft-lb.csh pw91 N
where pw91 indicates that the PW91 density functional was used, and N is the number of atoms in the cluster. - Now identify the unique structures with the command:
similarityAnalysis.py lb rotConstsData_C
where lb is used as a file-naming label. You now have a list of unique configurations optimized at the PW91/6-311++G** level of theory saved in the file uniqueStructures-lb.data.
- Compute the rotational constants of the large-basis-optimized clusters with the command:
5. Final Energy and Thermodynamic Correction Calculations
NOTE: The goal here is to obtain the vibrational structure and energies of the Gly(H2O)n=1-5 clusters using a large basis set and an ultrafine integration grid in order to compute the desired thermochemical corrections.
- Starting with results from the previous step, submit more reliable calculations.
- Create a subdirectory called ultrafine under the QM/pw91-lb directory. Then copy the unique structures list (uniqueStructures-lb.data) from the QM/pw91-lb directory to the QM/pw91-lb/ultrafine directory and change to that directory.
- Submit the ultrafine large-basis DFT script with the command:
run-pw91-lb-ultrafine.csh uniqueStructures-lb.data uf QUEUE 10
where uf is a label for this set of calculations, QUEUE is the preferred queue on the computing cluster, and 10 indicates that 10 calculations are to be grouped into one batch job. This script will automatically generate the inputs for Gaussian 09 and submit all the calculations. Enter 'test' for the 'QUEUE' to do a dry run testing.
- Once the submitted calculations are complete, extract and analyze the data
- Extract the energies and compute the rotational constants of the large-basis-optimized clusters with the command:
getRotConsts-dft-lb-ultrafine.csh pw91 N
where pw91 indicates that the PW91 density functional was used, and N is the number of atoms in the cluster. - Now identify the unique structures with the command:
similarityAnalysis.py uf rotConstsData_C
where uf is used as a file-naming label. You now have a list of unique configurations optimized at the PW91/6-311++G** level of theory saved in the file uniqueStructures-uf.data.
- Extract the energies and compute the rotational constants of the large-basis-optimized clusters with the command:
- Perform a final extraction of information needed to calculate thermodynamic corrections. Use that information to compute the thermodynamic corrections.
- Extract the final electronic energies, rotational constants and vibrational frequencies, and use them to calculate thermodynamic corrections using the command:
run-thermo-pw91.csh uniqueStructures-uf.data - Copy/paste the command-line output to the 'Raw_Energies' sheet of the Excel spreadsheet named 'gly-h2o-n.xlsx'. You would need to do this for the monomers (glycine and water) as well as the lowest energy member of each hydrate (gly-h2o-n, where n=1,2, …).
- As the raw energies are added to the first sheet of the 'gly-h2o-n.xlsx' spreadsheet, the subsequent 'Binding_Energies' and 'Hydrate_Distribution' sheets are automatically updated. In particular, the 'Hydrate_Distribution' sheet yields the equilibrium concentration of hydrates at different temperatures (Eg. 298.15K), relative humidity (20%, 50%, 100%) and initial concentrations of water ([H2O]) and glycine ([Glycine]). The theory behind these calculations is described in the next step.
- Extract the final electronic energies, rotational constants and vibrational frequencies, and use them to calculate thermodynamic corrections using the command:
6. Computing atmospheric concentrations of Gly(H2O)n=0-5 clusters at room temperature at sea-level
NOTE: This is accomplished by first copying the thermodynamic data generated in the previous step into a spreadsheet and calculating the Gibbs free energies of sequential hydration. Then, the Gibbs free energies are used to calculate equilibrium constants for each sequential hydration. Finally, a set of linear equations are solved to get the equilibrium concentration of the hydrates for a given concentration of monomers, temperature and pressure.
- Start by setting up a system of chemical equilibria for the sequential hydration of glycine as shown below:
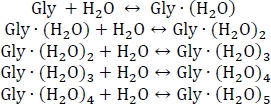
- Compute the equilibrium constants Kn using Kn = e-ΔGn/(kBT), where n is the level of hydration, ΔGn is the Gibbs free energy change of the nth hydration reaction, kB is Boltzmann's constant, and T is temperature.
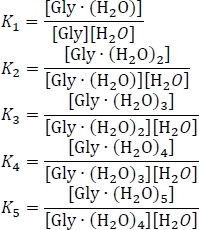
- Set up the equation for the conservation of mass, using the assumption that the sum of the equilibrium concentrations of the hydrated and un-hydrated glycine clusters equals the initial concentration of isolated glycine [Gly]0. Rewrite this system of six simultaneous equations, using some algebraic rearrangement of the equilibrium constant expressions, as
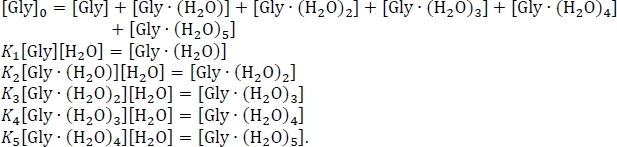
- Solve the system of equations shown above to obtain the equilibrium concentrations of Gly(H2O)n = 0-5 using an experimental value56,57,58 for the concentration of glycine in the atmosphere, [Gly]0 = 2.9 x 106 cm-3, and the concentration of water in the atmosphere at 100% relative humidity and a temperature of 298.15 K59, [H2O] = 7.7 x 1017 cm-3.
The first set of results from this protocol should be a set of low-energy structures of Gly(H2O)n=1-5 found through the configurational sampling procedure. These structures have been optimized at the PW91/6-311++G** level of theory and are assumed to be accurate for the purpose of this paper. There is no evidence to suggest that PW91/6-311++G** consistently underestimates or overestimates the binding energy of these clusters. Its ability to predict binding energies relative to MP2/CBS32 and [DLPNO-]CCSD(T)/CBS60,61 estimates and experiment52 shows a lot of fluctuations. The same is true of most other density functionals. Generally, each value of n = 1 – 5 should yield a handful of low-energy structures within around 5 kcal mol-1 of the lowest-energy structure. Here, we focus on the first structure produced by the run-thermo-pw91.csh script for brevity. Figure 3 shows the lowest electronic energy isomers of Gly(H2O)n=0-5 clusters. One can see that the hydrogen bond network grows in complexity as the number of water molecules increases, and even goes from a mostly planar network to a three-dimensional cage-like structure at n = 5. The rest of this text uses the energies and thermodynamic quantities corresponding to these five specific clusters.
Table 1 contains the thermodynamic quantities necessary to carry out the protocol. Table 2 shows an example of the output of the run-thermo-pw91.csh script where the electronic energies, vibrational zero-point corrections, and the thermodynamic corrections at three different temperatures are printed. For each cluster (row), E[PW91/6-311++G**] corresponds to the gas phase electronic energies at the PW91/6-311++G** level of theory calculated on ultrafine integration grids in units of Hartree, as well as the zero-point vibrational energy (ZPVE) in units of kcal mol-1. At each temperature, 216.65 K, 273.15 K, and 298.15 K, the thermodynamic corrections are listed, ∆H the enthalpy of formation in units of kcal mol-1, S the entropy of formation in units of cal mol-1, and ∆G the Gibbs free energy of formation in units of kcal mol-1. Table 3 shows an example computation of the total Gibbs free energy change of hydration, as well as for sequential hydration. An example computation of the total Gibbs free energy change of hydration for the reaction
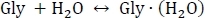
starts with the computation of the electronic energy EPW91 as
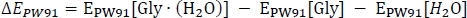
where EPW91[Gly∙(H2O)] is taken from Table 2 column C, and EPW91[Gly] and EPW91[H2O] are taken from Table 1 column B. Next we calculate the total gas phase energy change ΔE(0) by including the change in the zero-point vibrational energy of the reaction as
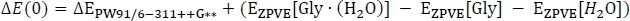
to obtain column D. Here, ΔEPW91/6−311++G** is taken from Table 3 column C, EZPVE[Gly ∙ (H2O)] from Table 2 column D, and EZPVE[Gly] and EZPVE[H2O] from Table 1 column C. For the sake of brevity, we will move on to room temperature clusters, so we skip over the 216.65 K and 273.15 K data. At room temperature, we then calculate the enthalpy change of the reaction ΔH by correcting the gas phase energy change as
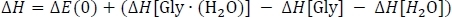
where ΔE(0) is taken from Table 3 column D, ΔH[Gly∙(H2O)] is taken from Table 2 column K, and ΔH[Gly] and ΔH[H2O] are taken from Table 1 column J. Finally, we calculate the Gibbs free energy change of the reaction ΔG as
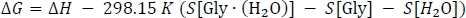
where ΔH is taken from Table 3 column I, S[Gly∙(H2O)] is taken from Table 2 column L, and S[Gly] and S[H2O] are taken from Table 1 column K. Note here that the entropy values must be converted to units of kcal mol-1 K-1 during this step.
We now have the necessary quantities to compute the atmospheric concentrations of hydrated glycine as shown in Step 6. The results should resemble the data shown in Table 4, but small numerical differences are to be expected. Table 4 shows the equilibrium hydrate concentrations found from the formulation of the system of six equations in Step 6.2 into one matrix equation and its subsequent solution. We start by acknowledging the fact that the system of equations can be written as
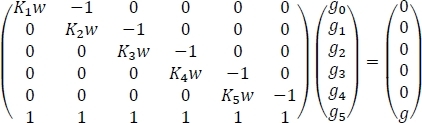
where Kn is the equilibrium constant for the nth sequential hydration of glycine, w is the concentration of water in the atmosphere, g is the initial concentration of isolated glycine in the atmosphere, and gn is the equilibrium concentration of Gly(H2O)n. If we rewrite the above equation as Ax = b, we get x = A−1b where A−1 is the inverse of matrix A. This inverse can be easily computed using built-in spreadsheet functions as shown in Table 4 to obtain the final results.
Figure 4 shows the equilibrium concentration of hydrated glycine calculated in Table 4 as a function of temperature at 100% relative humidity and 1 atmosphere pressure. It shows that, as temperature decreases from 298.15K to 216.65K, the concentration of unhydrated glycine (n=0) decreases and those of hydrated glycine increases. The glycine dihydrate (n=2) in particular increases dramatically with decreasing temperature while the change in the concentration of other hydrates is less noticeable. These inverse correlation between temperature and hydrate concentration is consistent with the expectation that lower Gibbs free energies of hydrations at lower temperatures favor the formation of hydrates.
Figure 5 illustrates the relative humidity dependence of equilibrium concentration of glycine hydrates at 298.15K and 1 atmosphere pressure. It clearly demonstrates that as RH increases from 20% to 100%, the concentration of hydrates (n>0) increase at the expense of unhydrated glycine (n=0). Once again the direct correlation between the relative humidity and concentration of hydrates is consistent with the idea that the presence of more water molecules at higher RH promotes the formation of hydrates.
As presented, this protocol gives a qualitative understanding of the hydrated glycine populations in the atmosphere. Assuming an initial concentration of isolated glycine of 2.9 million molecules per cubic centimeter, we see that the unhydrated glycine (n=0) is the most abundant species under most conditions except T=216.65K and RH=100%. The dihydrate (n=2), which has the lowest sequential Gibbs free energy of hydration at all three temperatures, is the most abundant hydrate at the conditions considered here. The monohydrate (n=1) and larger hydrates (n≥3) are predicted to be found in negligible amounts. Upon inspection of Figure 3, the abundance of the n = 1–4 clusters can be related to the stability and strain in the hydrogen bond network of the clusters. These clusters have the water molecules hydrogen bonded to the carboxylic acid moiety of glycine in a geometry closely resembling those of various hydrogen-bonded ring structures, making them especially stable.
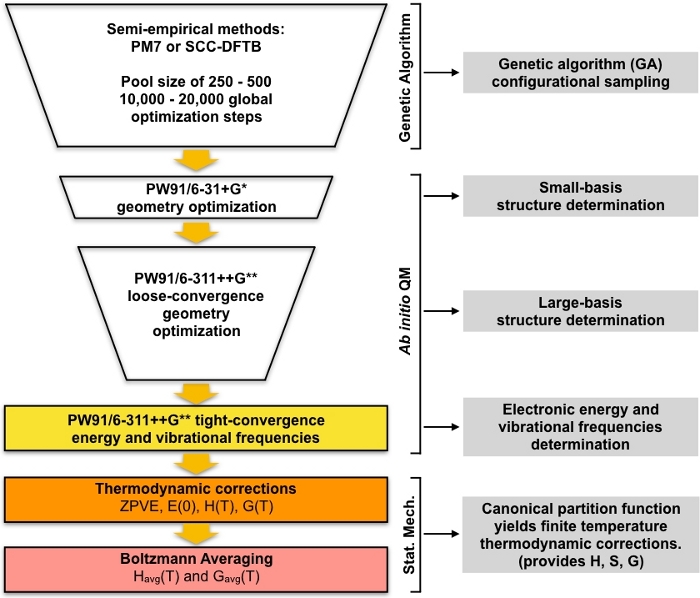
Figure 1: Schematic description of the current procedure. A large pool of guess structures generated by the genetic algorithm (GA) is refined by a series of PW91 geometry optimizations until a set of converged structures are obtained. The vibrational frequencies of these structures are computed and used to compute the Gibbs free energy of formation, which is in turn used to compute the equilibrium concentrations of the clusters under ambient conditions. Please click here to view a larger version of this figure.
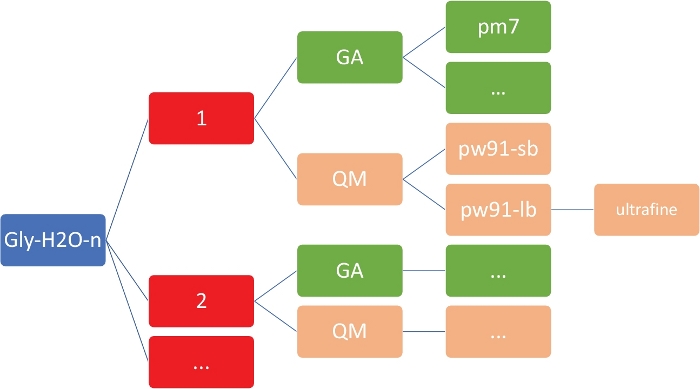
Figure 2: Representative directory structure for each cluster. The in-house scripts included in this protocol require the directory structure shown above, where n is the number of water molecules. For each n in gly-h2o-n, there are the following subdirectories: GA for genetic algorithm with a GA/pm7 directory, QM for quantum mechanics with QM/pw91-sb for PW91/6-31+G*, QM/pw91-lb for PW91/6-311++G**, and QM/pw91-lb/ultrafine for optimizations and final vibrational calculations on ultrafine integration grids. Please click here to view a larger version of this figure.
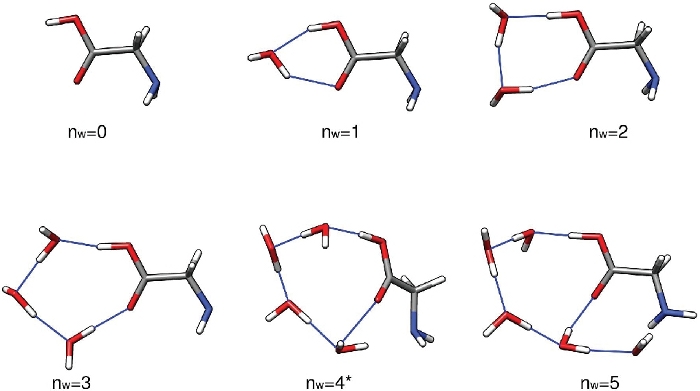
Figure 3: Representative low energy structures of Gly(H2O)n=0-5. These clusters were the electronic energy global minima optimized at the PW91/6-311++G** level of theory. Please click here to view a larger version of this figure.
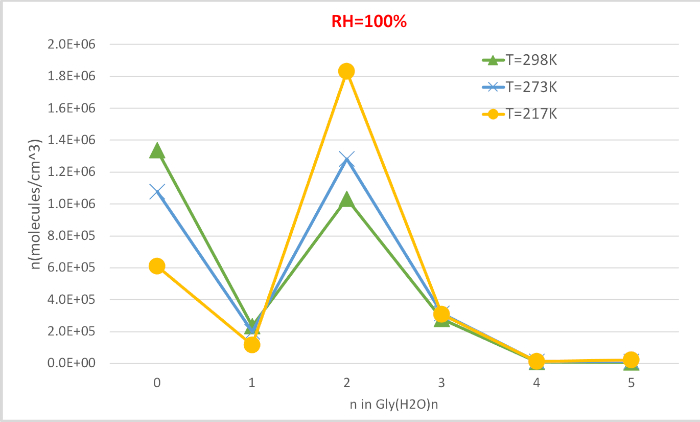
Figure 4: Temperature dependence of Gly(H2O)n=0-5 as 100% relative humidity and 1 atm pressure. The concentration of the hydrates is given in units of molecules cm-3. Please click here to view a larger version of this figure.
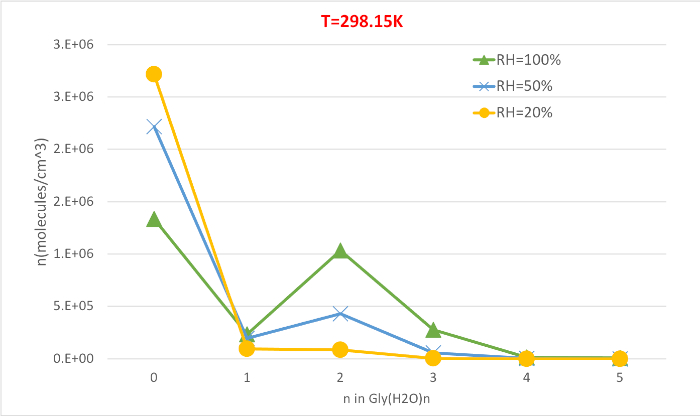
Figure 5: Relative humidity dependence of Gly(H2O)n=0-5 as 298.15 K and 1 atm pressure. The concentration of the hydrates is given in units of molecules cm-3. Please click here to view a larger version of this figure.
| E[PW91/6-311++G**] | 216.65 K | 273.15 K | 298.15 K | ||||||||
| LB-UF | ZPVE | ∆H | S | ∆G | ∆H | S | ∆G | ∆H | S | ∆G | |
| water | -76.430500 | 13.04 | 1.72 | 42.59 | 5.54 | 2.17 | 44.44 | 3.08 | 2.37 | 45.14 | 1.96 |
| glycine | -284.434838 | 48.55 | 2.65 | 69.53 | 36.14 | 3.70 | 73.81 | 32.09 | 4.22 | 75.61 | 30.22 |
Table 1: Monomer energies. Electronic energies are in units of Hartree while all other quantities are in units of kcal mol-1. Water and glycine were optimized at the PW91/6-311++G** level of theory and vibrational frequencies were computed. The thermodynamic corrections for a pressure of 1 atm and temperature of 298.15 K were computed using the thermo.pl script.
| E[PW91/6-311++G**] | 0 K | 216.65 K | 273.15 K | 298.15 K | ||||||||
| n | nome | LB-UF | ZPVE | ∆H | S | ∆G | ∆H | S | ∆G | ∆H | S | ∆G |
| 1 | gly-h2o-1 | -360.88481 | 63.96 | 3.61 | 80.12 | 50.22 | 5.12 | 86.27 | 45.52 | 5.85 | 88.83 | 43.33 |
| 2 | gly-h2o-2 | -437.33763 | 79.33 | 4.53 | 90.86 | 64.17 | 6.46 | 98.78 | 58.81 | 7.40 | 102.06 | 56.30 |
| 3 | gly-h2o-3 | -513.78620 | 94.52 | 5.67 | 105.08 | 77.42 | 8.08 | 114.94 | 71.19 | 9.23 | 119.00 | 68.27 |
| 4 | gly-h2o-4 | -590.23667 | 109.80 | 6.03 | 104.98 | 91.30 | 8.78 | 116.21 | 84.40 | 10.11 | 120.87 | 81.14 |
| 5 | gly-h2o-5 | -666.68845 | 125.80 | 7.26 | 121.70 | 106.69 | 10.47 | 134.83 | 99.44 | 12.01 | 140.24 | 96.00 |
Table 2: Cluster energies. The energies of the lowest-energy Gly(H2O)n=1-5 structures found using our procedure outlined in Figure 1. Electronic energies are in units of Hartree while all other quantities are in units of kcal mol-1.
| Total Hydration: Gly + nH2O <-> Gly(H2O)n | Sequential Hydration: Gly(H2O)n-1 + H2O <-> Gly(H2O)n | ||||||||||||||||
| E[PW91/6-311++G**] | 216.65 | 273.15 | 298.15 | 216.65 | 273.15 | 298.15 | |||||||||||
| n | system name | LB-UF | ∆E(0) | ∆H(T) | ∆G(T) | ∆H(T) | ∆G(T) | ∆H(T) | ∆G(T) | LB-UF | ∆E(0) | ∆H(T) | ∆G(T) | H(T) | ∆G(T) | ∆H(T) | ∆G(T) |
| 1 | gly-h2o-1 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 |
| 2 | gly-h2o-2 | -26.22 | -21.53 | -23.10 | -9.27 | -23.11 | -5.66 | -23.09 | -4.06 | -14.00 | -11.68 | -12.49 | -5.59 | -12.50 | -3.79 | -12.50 | -2.99 |
| 3 | gly-h2o-3 | -37.56 | -30.72 | -32.88 | -12.90 | -32.87 | -7.69 | -32.82 | -5.38 | -11.34 | -9.19 | -9.78 | -3.63 | -9.76 | -2.03 | -9.73 | -1.32 |
| 4 | gly-h2o-4 | -50.10 | -40.34 | -43.48 | -15.87 | -43.54 | -8.71 | -43.51 | -5.55 | -12.54 | -9.62 | -10.60 | -2.97 | -10.67 | -1.02 | -10.69 | -0.17 |
| 5 | gly-h2o-5 | -63.45 | -51.41 | -55.42 | -20.58 | -55.51 | -11.48 | -55.48 | -7.45 | -13.35 | -11.07 | -11.94 | -4.71 | -11.97 | -2.77 | -11.97 | -1.90 |
Table 3: Hydration energies. The total energy of hydration and energy of sequential hydration for Gly(H2O)n=1-5 in units of kcal mol-1. Here, E[PW91/6-311++G**] is the change in the electronic energy, ∆E(0) is the zero-point vibrational energy (ZPVE) corrected change in energy, ∆H(T) is the enthalpy change at temperature T, and ∆G(T) is the Gibbs free energy change of hydration of each Gly(H2O)n=1-5 cluster.
| Equilibrium Hydrate Distribution as a function of temperature and relative humidity | |||||||||
| T=298.15K | T=273.15K | T=216.65K | |||||||
| Gly(H2O)n | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% |
| 0 | 1.3E+06 | 2.2E+06 | 2.7E+06 | 1.1E+06 | 2.0E+06 | 2.7E+06 | 6.1E+05 | 1.5E+06 | 2.5E+06 |
| 1 | 2.3E+05 | 1.9E+05 | 9.5E+04 | 2.0E+05 | 1.9E+05 | 9.9E+04 | 1.2E+05 | 1.5E+05 | 9.5E+04 |
| 2 | 1.0E+06 | 4.3E+05 | 8.4E+04 | 1.3E+06 | 6.1E+05 | 1.3E+05 | 1.8E+06 | 1.1E+06 | 3.0E+05 |
| 3 | 2.8E+05 | 5.8E+04 | 4.5E+03 | 3.2E+05 | 7.4E+04 | 6.3E+03 | 3.1E+05 | 9.6E+04 | 1.0E+04 |
| 4 | 1.1E+04 | 1.1E+03 | 3.4E+01 | 1.3E+04 | 1.5E+03 | 5.0E+01 | 1.1E+04 | 1.8E+03 | 7.5E+01 |
| 5 | 7.5E+03 | 3.9E+02 | 4.9E+00 | 1.2E+04 | 7.2E+02 | 9.7E+00 | 2.4E+04 | 1.9E+03 | 3.1E+01 |
Table 4: Equilibrium hydrate concentrations of Gly(H2O)n=0-5 as a function temperature (T=298.15K, 273.15K, 216.65K) and relative humidity (RH=100%, 50%, 20%). The concentration of the hydrates is given in units of molecules cm-3 assuming experimental values56,57,58, of [Gly]0 = 2.9 x 106 cm-3 and [H2O] = 7.7 x 1017 cm-3, 1.6 x 1017 cm-3 and 9.9 x 1014 cm-3 at 100% relative humidity and T = 298.15 K, 273.15 K, and 216.65 K, respectively59.
Supplemental Files. Please click here to download these files.
