Toxoplasma gondii is categorized in Risk Group 2 and must be handled at a Biosafety Level 2 (BSL-2). The protocol has been reviewed and approved by the Institutional Biosafety Committee at Clemson University.
1. Luciferase-based Toxoplasma growth assay
- Seed human foreskin fibroblasts (HFFs) 1 week before parasite inoculation to ensure that host cells are fully confluent. Perform a mock assay in a transparent plate to ensure that parasites remain intracellular throughout the evaluation period.
NOTE: Here, the assay is conducted in 96 well microplates. According to experimental needs, it can be scaled up to 384 or 1536 well microplates. - Pass Toxoplasma parasites into confluent HFFs 2 days prior to use by transferring ~0.3-0.4 mL of fully lysed parasites into a T25 flask. Incubate infected host cells at 37 °C with 5% CO2 for 2 days.
- Syringe 5 mL of freshly lysed parasites through a 21 G safety needle 5x to liberate intracellular parasites, then pass through a 3 µm filter to remove host cell debris. Rinse residual parasites out of the flask using 7 mL of phenol red-free D10 medium, then pass through the filter again.
- Centrifuge parasites at 1000 x g for 10 min at room temperature (RT). Pour off the supernatant and resuspend the pellet in 10 mL of phenol red-free D10 media.
- Count parasites using a hemocytometer to determine the concentration.
- Dilute parasites to 1 x 104 parasites/mL for the wild-type (WT) strain. For growth-deficient parasite strains, increase the concentration accordingly to observe a significant increase in luciferase signals.
- Aspirate media carefully from 96 well microplates pre-seeded with HFFs and inoculate 150 µL of parasite resuspension into wells in a format of three columns and five rows, which represents three technical replicates and five timepoints.
- Incubate the microplate at 37 °C and 5% CO2 for 4 h.
- Aspirate media carefully from the wells to remove non-invaded parasites, then fill the wells with RT phenol red-free media in each row (except for the first row).
- Mix equal volumes of PBS and 2x luciferase assay buffer and dilute the luciferase substrate to 12.5 µM.
- Add 100 µL of dilute luciferase substrate into each well of the top row. Incubate the microplates at RT for 10 min to allow the cells to fully lyse.
- Measure the luciferase activity using a microplate reader. The plate reader settings are listed in Table 1. Each reading represents the initial number of invaded parasites at 4 h post-infection.
- Repeat steps 1.9-1.12 for each row every 24 h for 4 days without changing the medium. These readings reflect the total number of replicated parasites at 24 h, 48 h, 72 h, and 96 h post-infection.
- Calculate the average readings at each timepoint and divide them by the average readings at 4 h to determine the fold changes in parasite growth over time.
- Plot the data using graphing software. A representative growth reading table and plots of RHΔku80::NLuc parasites are shown in Figure 1A,B.
- To calculate doubling time, plot the log2 values of fold changes at the individual timepoints over the incubation time. Use a linear regression function to calculate slope, which represents the doubling time of each strain (Figure 1A,C).
2. Evaluation of chemical compound inhibition efficacy against Toxoplasma growth
NOTE: Here, evaluation of the inhibition of LHVS in Toxoplasma growth is presented as an example. Eight different concentrations of LHVS are tested, and three technical replicates are performed for each of the three biological replicates for both RHΔku80::NLuc and RHΔku80Δcpl::NLuc strains.
- Prior to the parasite infection, seed HFFs to 96 well microplates in the format of three rows and nine columns for one biological replicate per compound per strain. Host cells will be allowed to grow for at least 7 days before use.
- Pass RHΔku80::NLuc and RHΔku80Δcpl::NLuc parasites for 2 days prior to use. Follow steps 1.2-1.6 for parasite purification and quantification. Resuspend parasites in phenol red-free media at 1 x 104 parasites/mL.
- Aspirate media from a plate containing confluent HFFs and inoculate each well with 150 µL of parasite resuspension. Incubate the microplate at 37 °C and 5% CO2 for 4 h.
- Prepare LHVS at eight different concentrations in a 12 well reservoir by serial dilution. Generally, the concentrations are decreased by three-fold in a serial dilution manner.
NOTE: The lowest concentration is reduced by 6,561-fold relative to the highest concentration. The fold change of the dilution can be adjusted accordingly based on different properties of individual compounds. - At 4 h post-infection, aspirate media to remove non-invaded parasites and fill each well from columns 2-9 with 150 µL of media supplemented with LHVS at different concentrations. Leave the first column filled with regular medium to serve as a nontreated control.
- Incubate the microplate at 37 °C and 5% CO2 for an additional 96 h.
- Perform steps 1.9-1.11 and measure luciferase activity of individual wells.
- Average the luciferase activities of three technical replicates from wells of each individual LHVS concentration.
- Divide the average luciferase activity for each LHVS concentration by the average luciferase activity derived from nontreated parasites to calculate the normalized luciferase activity as a percentage.
- Plot the normalized luciferase activities against the individual LHVS concentrations using graphing software (Figure 2). Inhibition of pyrimethamine against parasite growth is also measured as a control. Pyrimethamine is a clinical antibiotic used to treat acute toxoplasmosis by inhibiting folic acid metabolism in Toxoplasma.
- Calculate the IC50 values for individual compounds using the embedded method in the graphing software, normalized response vs. [inhibitor], under the "dose-response-inhibition" regression program. The IC50 is calculated using the following formula:
Y = 100/(1 + X/IC50)
Where: Y represents normalized luciferase activities of infected cells under different concentrations of inhibitor, and X represents individual concentrations of inhibitor.
3. CRISPR-Cas9-based gene deletion in Toxoplasma parasites
- Generation of a plasmid construct expressing guide RNA (sgRNA) and Cas9 for deleting a gene of interest
- Go to www.ToxoDB.org and retrieve the entire gene coding sequence, including introns and exons, along with 1.5 kb 5'-UTRs and 3'-UTRs (untranslated regions).
NOTE: Here, TgCPL (TGGT1_321530) is targeted as a representative example. - Copy the retrieved TgCPL sequence into the sequence analysis software (refer to Table of Materials for the name and version) and label the 5'- and 3'-UTR regions.
- Select the Tools icon in the top menu bar, then select Cloning | Find CRISPR Sites.
- Choose 3'(Cas9)' for the PAM site location and select the folder containing the Toxoplasma genome sequence in the specificity scoring section. Leave the rest of the settings as defaults.
- Choose a sgRNA with the following two criteria: 1) showing a high specificity score, generally >98%, and 2) lacking a G following the NGG, a protospacer adjacent motif (PAM) sequence. The selected sgRNA is usually located at sites close to the start and stop codons of the gene of interest.
- Copy the sequence of the selected sgRNA and paste it into the following primer template.

The portion in red represents the selected TgCPL sgRNA sequence. It can be replaced with different sgRNAs for various genes of interest.
NOTE: If the selected sgRNA does not start with G, add G at the beginning of the sgRNA to help enhance its expression. - Perform a PCR reaction to modify the pre-existing plasmid expressing sgRNA (Figure 3A) that targets Toxoplasma uracil phosphoribosyltransferase (TgUPRT) gene23 using a PCR premix with the settings provided in Table 2.
- Run the PCR product on an agarose gel to confirm successful amplification. A 10 kb PCR product is expected to be amplified (Figure 3B).
- Extract the PCR product using a DNA gel extraction kit and circularize it using a site-directed mutagenesis kit. Refer to Table 3 for the recipe. Incubate the reaction for 10-20 min at RT.
- Transform the circularized PCR product into E. coli and pick 10 clones for further verification of incorporation of designed sgRNA.
- Grow two clones and extract plasmids. Cut the purified plasmids with BamHI and EcoRV. The candidate plasmids will yield two bands at 2.4 kb and 7.2 kb (Figure 3C).
- Send the plasmids for Sanger sequencing using M13 reverse primers to confirm successful replacement of TgUPRT sgRNA with the designed sgRNA (Figure 3D).
- Go to www.ToxoDB.org and retrieve the entire gene coding sequence, including introns and exons, along with 1.5 kb 5'-UTRs and 3'-UTRs (untranslated regions).
- Generation of repair template for gene deletion via HDR mechanism
- According to the targeting sites of the selected sgRNA, locate 50 bp of 5'-UTRs or 3'-UTRs of the target gene for homology-dependent recombination (HDR, see discussion section). The selection of regions follows the criteria listed below, depending on the location the sgRNA targets.
- If the cleavage site by Cas9 is located upstream from the start codon, select the following: a 50 bp DNA sequence upstream from the cleavage site as the left HDR region, and a 50 bp DNA sequence downstream from the stop codon as the right HDR region.
- If the cleavage site by Cas9 is between the start and stop codons, select the following: a 50 bp DNA sequence upstream from the start codon as the left HDR region, and a 50 bp DNA sequence downstream from the stop codon as the right HDR region.
- If the cleavage site by Cas9 is located downstream from the stop codon, select the following: a 50 bp DNA sequence upstream from the start codon as the left HDR region, and a 50 bp DNA sequence downstream from the cleavage site as the right HDR region.
NOTE: For the TgCPL gene, the cleavage site is located between the start and stop codons. Thus, the following primers are designed for amplifying the repair template using pMDC64 as the template, which encodes a pyrimethamine resistance cassette. The sequences in black anneal to the pMDC64 plasmid for PCR amplification. The regions labeled in red are TgCPL-specific sequences for homologous recombination.

- Perform PCR using a PCR premix under the PCR conditions described in Table 4.
- Run the PCR product on an agarose gel (Figure 3E), followed by gel extraction and standard nucleic acid quantification procedures.
NOTE: If the expected band cannot be successfully amplified, optimize PCR conditions and/or switch primer pairs.
- According to the targeting sites of the selected sgRNA, locate 50 bp of 5'-UTRs or 3'-UTRs of the target gene for homology-dependent recombination (HDR, see discussion section). The selection of regions follows the criteria listed below, depending on the location the sgRNA targets.
- Toxoplasma transfection
- Pass RHΔku80::NLuc parasites for 2 days in a T25 flask containing confluent HFFs. A T25 flask of fully lysed parasites is sufficient for two to three transfections.
- Syringe and filter-purify parasites as described in step 1.2. Resuspend parasites in cytomix buffer and spin down at 1,000 x g for 10 min at RT.
- Wash pelleted parasites with 10 mL of cytomix buffer and spin down the parasites at 1,000 x g for 10 min at RT.
- Carefully pour off the supernatant and resuspend the parasites in the same buffer at a concentration of 1 x 108 parasites/mL.
- Mix 2 µg of repair template DNA with 20 µg of the sgRNA/Cas9 expression plasmids (mass ratio = 1:5, equivalent to a 1:3 molar ratio). If the amplification yield of repair template is low, reduce the input of both DNA pieces accordingly. A minimum of 0.5 µg of repair template can be used.
- Mix 400 µL of parasite resuspension, DNA, and 5 µL of 200 mM ATP/500 mM reduced glutathione (GSH) in a 1.5 mL centrifuge tube. Bring the total volume to 500 µL with cytomix buffer, if needed.
- Transfer the mixture of parasites and DNA to an electroporation cuvette (4 mm gap width) and perform electroporation (2 kV voltage, 50 Ω resistance) using an electroporation apparatus.
- Transfer electroporated parasites to a T25 flask containing confluent HFFs in fresh D10 medium. Apply appropriate antibiotic for drug selection after 24 h.
- Keep drug selective pressure until the growth of the transgenic parasites is stable.
- Purify genomic DNA from the knockout population and check for integration of the pyrimethamine resistance cassette into the TgCPL locus by PCR. After verified, proceed to section 3.4. If not, perform another round of parasite transfection and drug selection. Inability to detect the correct integration of the drug resistance cassette usually suggests that the target gene is essential or that the gene locus is not accessible.
- Cloning of knockout parasites
- Seed two 96 well microplates with HFF cells and incubate at 37 °C and 5% CO2 for 1 week prior to cloning parasites.
- Pass ~0.3-0.4 mL of the population of transgenic parasites in a T25 flask containing confluent HFFs and grow them for 2 days. Consider passing more parasites if the mutant shows growth defects.
NOTE: To achieve the best yield and viability, the host cells are heavily infected by the parasites, and most of the parasites are kept in the intracellular stage. - Syringe infected host cells and filter-purify freshly lysed parasites as mentioned in step 1.3. Resuspend the parasites in D10 medium and spin them down at 1,000 x g for 10 min at RT.
- Resuspend the pelleted parasites in 10 mL of D10 medium.
- Count parasites using a hemocytometer to determine the parasite concentration.
- Conduct a two-step dilution to bring the concentration to 10 parasites/mL in D10 medium supplemented with the appropriate antibiotic. Usually, the initial parasite resuspension is diluted by 1,000-fold, followed by a second dilution to 10 parasites/mL.
- Aspirate media from 96 well microplates containing confluent HFFs and inoculate 150 µL of diluted parasites into each well.
- Incubate plates at 37 °C with 5% CO2 for 7 days without disturbance to allow plaque formation. The incubation period can be longer if transgenic parasites exhibit growth defects.
- Screen the plates using a phase-contrast microscope and mark only the wells containing a single plaque.
- Perform colony PCR to identify correct clones.
- Use pipette tips to scrape the bottom of each well to lift infected HFF monolayers.
- Pipet 75 µL of the cell resuspension from each marked well into 1.5 mL microcentrifuge tubes.
- Centrifuge tubes for 10 min at maximum speed at RT. Carefully aspirate the supernatant and resuspend the pellet in 10.25 µL of lysis buffer containing dilution buffer and DNA release additive provided in the kit (Table of Materials).
- Incubate the samples for 4 min at RT, then 2 min at 98 °C. Afterward, samples can be used for PCR or stored at -20 °C until use. Three sets of PCR reactions are used to test for the integration of the drug resistance cassette and loss of the gene of interest (Figure 4A). Refer to Table 5 for PCR reaction setup and Table 6 for thermocycler settings.
- Identify the correct clones and transfer four clones into T25 flasks containing confluent HFFs.
- After individual clones lyse host cells, purify genomic DNA for further PCR verification.
- If an antibody recognizing the protein of interest is available, follow a standard immunoblotting procedure to verify loss of the target protein in the correct Toxoplasma knockouts. Representative images for screening a TgCPL-deletion mutant are shown in Figure 4B,C.
Figure 1 represents an example of a growth curve for the RHΔku80::NLuc strain and the derived calculation for its doubling time. Generally, the assay is performed in three technical replicates for each of the three biological replicates to account for variations of luciferase activity readings. In order to calculate the normalized fold change of parasite growth, each reading at 24-96 h post-infection was divided by the initial reading at 4 h post-infection, which reflects the starting amount of live parasites in the assay (Figure 1A,B). In terms of determining parasite doubling time, the log2 values of the normalized fold changes of parasite growth were plotted against each timepoint. Next, the plot was subjected to a linear regression function to obtain the slope, which represents doubling time (Figure 1C).
The inhibition efficacies of LHVS in wild-type and Δcpl strains were determined by plotting luciferase activities against eight inhibitor concentrations in Figure 2. It is essential to include infected cells without inhibitor treatment for normalization of raw luciferase activities in the assay. In addition, a mock experiment performed in a clear microplate is required for the assay to ensure that parasites are still in the intracellular stage at the end of the assay period.
In Figure 3, the generation and validation of a sgRNA expression construct targeting TgCPL and the production of a repair template for TgCPL deletion are shown. The 20 bp sgRNA matching to the TgUPRT gene encoded in the original plasmid was mutated to the DNA sequence targeting the TgCPL gene via PCR-based site-directed mutagenesis. To achieve this, the DNA sequences coding for the sgRNAs that recognize different genes were engineered to the forward primer, while the reverse primer was kept unchanged to simplify primer design.
Figure 3A shows a zoomed-in region of the sgRNA DNA sequences targeting the TgUPRT gene in the original template plasmid as well as the primer set used for the generation of the linearized sgRNA expression vector. Figure 3B shows a representative gel picture of the linearized TgCPL-targeting sgRNA expression plasmid. Figure 3C shows the restriction endonuclease digestion of the circularized TgCPL-targeting sgRNA expression plasmid. A M13 reverse primer was used to sequence the incorporated guide RNA within the sgRNA expression vector generated for the specific gene. In Figure 3D, the sequenced DNA region was aligned to the plasmid template for the confirmation of successful mutagenesis. Figure 3E illustrates the start and end regions of the pyrimethamine resistance cassette, showing where the primers can anneal for production of the repair template for TgCPL gene deletion. The repair template was PCR-amplified and loaded into a 1% agarose gel for size verification and gel extraction.
The overall strategy for TgCPL knockout generation and screening is shown in Figure 4. Three sets of primers shown in Figure 4A were used to screen TgCPL-deletion parasites for the correct integration of 5'- and 3'-ARMs and deletion of the TgCPL-coding sequence. As shown in Figure 4B, generally, seven to eight clones are selected for screening initially. The screening usually starts with checking for deletion of the coding sequence for the gene of interest. This is followed by detection of 5' and 3'-ARMs, which helps minimize the total number of clones to be screened. Further verification by immunoblotting displayed in Figure 4C can be completed if an antibody recognizing the target protein is available.
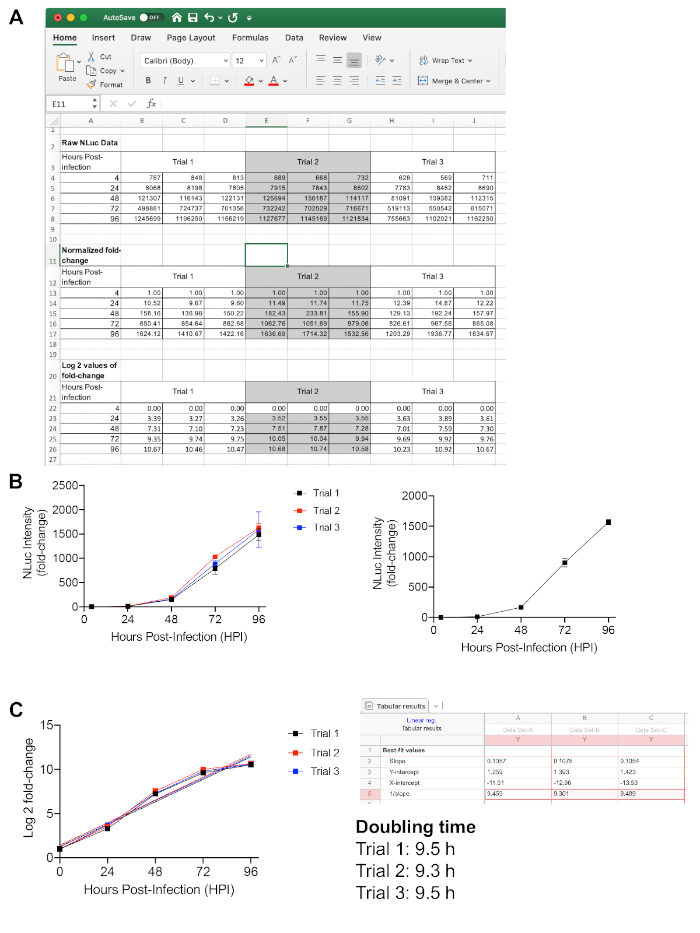
Figure 1: Intracellular growth quantification for Toxoplasma parasites using a luciferase-based method. (A) Raw luciferase activity readings in a spreadsheet software. The readings at 24 h, 48 h, 72 h, and 96 h post-infection were normalized against the initial readings at 4 h post-infection for calculating the fold changes in parasite growth. (B) The normalized data were averaged and plotted. (C) The log2 values of the fold changes were also plotted and subjected to linear regression for determination of the parasite's doubling time. Please click here to view a larger version of this figure.
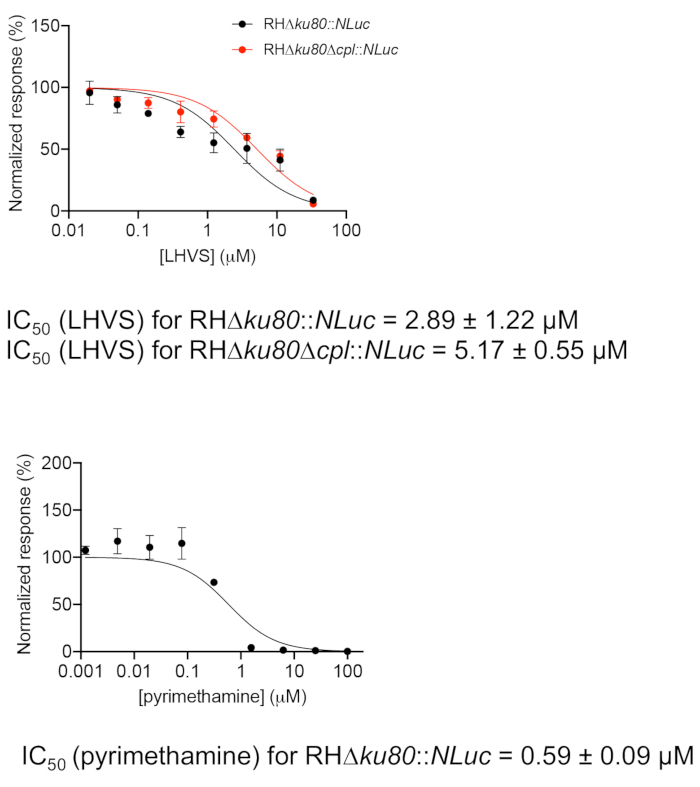
Figure 2: Inhibition efficacy assessment of LHVS and pyrimethamine using the luciferase-based growth assay. Parasites were inoculated into a 96 well microplate for 4 h to allow for invasion of host cells. Non-invaded parasites were washed away, and the plate was filled with media containing different concentrations of LHVS or pyrimethamine and incubated for an additional 96 h before determination of luciferase activity. The measured luciferase readings for parasites treated with individual inhibitor concentrations were normalized against the signal detected from untreated parasites. The data were plotted in a graphing program, and a regression analysis for IC50 determination was performed. The assay was repeated in three biological replicates with three technical replicates each. Data represent mean ± SEM, n = 3 biological replicates. Please click here to view a larger version of this figure.
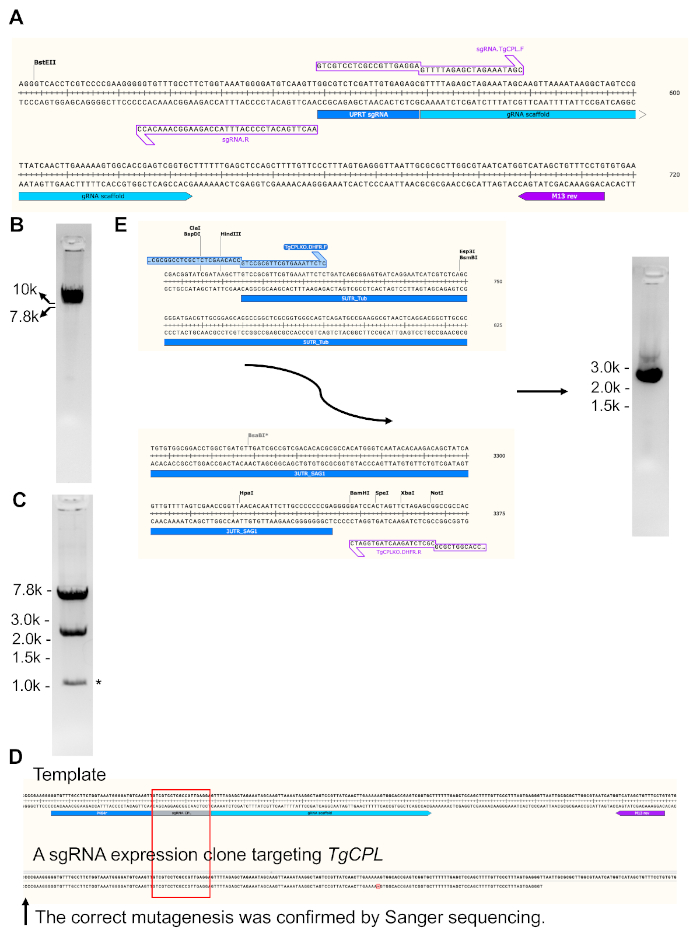
Figure 3: Generation of the plasmid construct expressing sgRNA targeting TgCPL and production of a repair template for TgCPL deletion. (A) The original pSAG1-Cas9-sgRNA-UPRT plasmid23 was modified via a site-directed mutagenesis kit for replacement of the sgRNA targeting the TgUPRT gene to TgCPL. The sgRNA coding region is enlarged to show areas to which the primers anneal. After PCR, the mutated plasmid was linearized and loaded into a 1% agarose gel for verification of successful amplification, followed by gel extraction. (B) The gel image of the PCR-amplified linearized sgRNA expression construct. (C) After gel-extraction, the PCR product was circularized and subsequently transformed into E. coli. The clones containing the expected plasmids were screened by restriction endonuclease digestion and DNA sequencing. The band sizes after DNA digestion were 7.2 bp and 2.4 kb. The band generated by nonspecific cleavage from endonucleases is labeled by asterisk. (D) The M13 reverse primer labeled in the figure was used to sequence the mutated guide RNA region within the generated TgCPL-targeting sgRNA expression vector. The sequenced DNA region was aligned to the plasmid template to confirm successful mutagenesis. (E) In this study, 50 bp homologous regions matching to the 5'- and 3'-UTRs of TgCPL were engineered into the primers for amplification of the repair template and flanked at the 5'- and 3'-ends of the pyrimethamine resistance cassette by PCR, respectively. Agarose gel electrophoresis was used to verify the correct size of the PCR product before gel extraction. The expected size of the repair template is ~2.7 kb. Usually, 5-6 µg of repair template can be obtained from 200 µL of PCR reaction. Please click here to view a larger version of this figure.
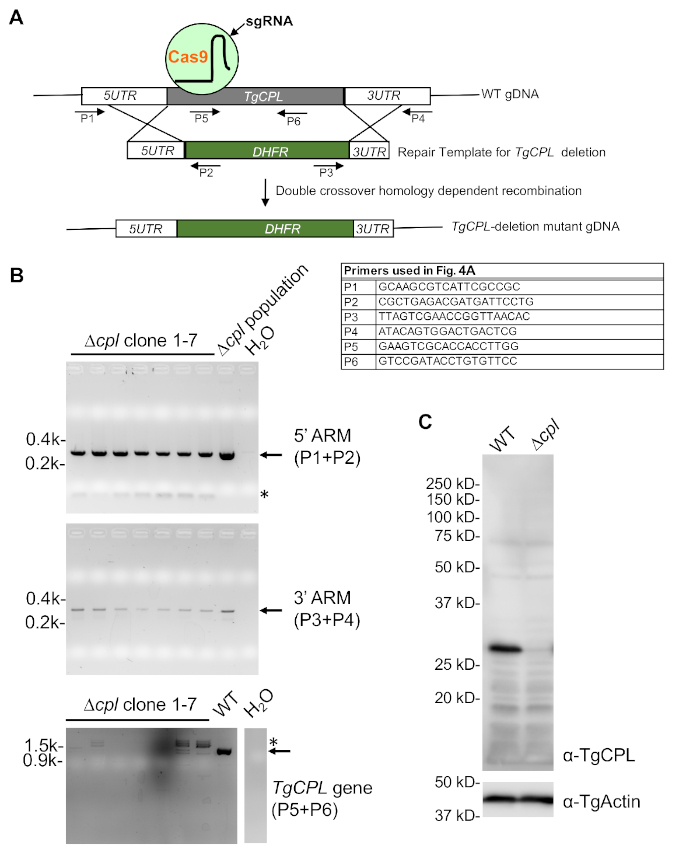
Figure 4: PCR and immunoblotting confirmation of TgCPL-deficient parasites. (A) A schematic diagram depicting the general strategies of TgCPL-deletion in Toxoplasma and PCR-based screening of the correct TgCPL knockout clones. The primers used for the screening are labeled. (B) PCR and agarose gel electrophoresis were used to select clones containing the correct integration of the pyrimethamine resistance cassette into the TgCPL locus and loss of the TgCPL gene. The genomic DNA of the Δcpl population served as a positive control for 5'- and 3'-ARM detection, while the WT genomic DNA was used for the detection of the TgCPL gene as a positive control. Water was used instead of DNA template in the PCR reactions to serve as a negative control. The expected bands are denoted by arrows, whereas nonspecific PCR amplifications are labeled by asterisks. (C) Clone 1 identified by PCR screening was grown in tissue culture for cell lysate preparation and further immunoblotting analysis to confirm the loss of TgCPL expression in the knockout. TgActin was used as a loading control. Please click here to view a larger version of this figure.
| Luciferase: | Endpoint |
| Integration time: | 1 s |
| Filter Set – Emission: | Full light |
| Optics: | Top |
| Gain: | 135 |
| Read speed: | Normal |
| Delay: | 100 ms |
| Read height: | 4.5 mm |
Table 1: Microplate reader settings for luciferase activity measurement during luciferase-based Toxoplasma growth assay.
| Initial denaturation: | 98 °C for 5 min |
| 25 cycles of | |
| Denaturing: | 98 °C for 5 s |
| Annealing: | 60 °C for 15 s |
| Extension: | 72 °C for 1 min |
| Final extension: | 72 °C for 10 min |
Table 2: Thermocycler settings for generation of sgRNA expression vector.
| Sample | Volume (µl) |
| PCR product (10-50 ng) | 1 |
| 2X KLD (kinase, ligase, DpnI) Reaction Buffer | 5 |
| 10X KLD Enzyme Mix | 1 |
| Nuclease-free water | 3 |
| Total | 10 |
Table 3: Reaction recipe for circularization of sgRNA expression vector.
| Initial denaturation: | 98 °C for 5 min |
| 35 cycles of | |
| Denaturing: | 98 °C for 15 s |
| Annealing: | 58 °C for 15 s |
| Extension: | 72 °C for 30 s per kb |
| Final extension: | 72 °C for 10 min |
Table 4: Thermocycler setting for generation of repair template.
| Sample | Volume (µl) |
| total Toxoplasma genomic DNA | 1 |
| Forward primer (25 µM) | 0.2 |
| Reverse primer (25 µM) | 0.2 |
| 2x PCR master premix | 5 |
| Nuclease-free water | 3.6 |
| Total | 10 |
Table 5: Colony PCR reaction recipe for screening single Toxoplasma clones.
| Initial denaturation: | 98 °C for 5 min |
| 35 cycles of | |
| Denaturing: | 98 °C for 5 s |
| Annealing: | 55 – 62 °C for 5 s |
| Extension: | 72 °C for 20 s per kb |
| Final extension: | 72 °C for 1 min |
Table 6: Thermocycler setting for screening single Toxoplasma clones.
| Cytomix buffer | 25 mM HEPES, pH 7.6, 120 mM KCl, 10 mM K2HPO4/KH2PO4, 5 mM MgCl2, 0.015 mM CaCl2, and 2 mM EGTA. |
| D10 medium | DMEM 1X (Corning, Cat #: 10-013-CV), 10 mM HEPES, 10% (v/v) Cosmic Calf Serum (Hyclone, Cat #: SH30087.03), 1 mM sodium pyruvate, 4 mM L-glutamine, 100 units/mL of penicillin, and 100 µg/mL of streptomycin. |
| Phenol red-free medium | DMEM/ Highly Modified (Hyclone, Cat #: SH30284.02), 10 mM HEPES, 10% (v/v) Cosmic Calf Serum (Hyclone, Cat #: SH30087.03), 1 mM sodium pyruvate, 4 mM L-glutamine, 100 units/mL of penicillin, and 100 µg/mL of streptomycin. |
| 2X NLuc Buffer | 100 mM MES, pH 6.0, 1mM CDTA, 0.5% Tergitol, 0.05% Mazu DF 204, 150 mM KCl, 1 mM DTT, 35 mM Thiourea. |
Supplementary Table 1: Recipes for buffers.
