Assessing the Expression of Major Histocompatibility Complex Class I on Primary Murine Hippocampal Neurons by Flow Cytometry
Summary
This protocol describes a method for culturing primary hippocampal neurons from embryonic mouse brain. The expression of major histocompatibility complex class I on the extracellular surface of cultured neurons is then assessed by flow cytometric analysis.
Abstract
Increasing evidence supports the hypothesis that neuro-immune interactions impact nervous system function in both homeostatic and pathologic conditions. A well-studied function of major histocompatibility complex class I (MHCI) is the presentation of cell-derived peptides to the adaptive immune system, particularly in response to infection. More recently it has been shown that the expression of MHCI molecules on neurons can modulate activity-dependent changes in the synaptic connectivity during normal development and neurologic disorders. The importance of these functions to the brain health supports the need for a sensitive assay that readily detects MHCI expression on neurons. Here we describe a method for primary culture of murine hippocampal neurons and then assessment of MHCI expression by flow cytometric analysis. Murine hippocampus is microdissected from prenatal mouse pups at the embryonic day 18. Tissue is dissociated into a single cell suspension using enzymatic and mechanical techniques, then cultured in a serum-free media that limits growth of non-neuronal cells. After 7 days in vitro, MHCI expression is stimulated by treating cultured cells pharmacologically with beta interferon. MHCI molecules are labeled in situ with a fluorescently tagged antibody, then cells are non-enzymatically dissociated into a single cell suspension. To confirm the neuronal identity, cells are fixed with paraformaldehyde, permeabilized, and labeled with a fluorescently tagged antibody that recognizes neuronal nuclear antigen NeuN. MHCI expression is then quantified on neurons by flow cytometric analysis. Neuronal cultures can easily be manipulated by either genetic modifications or pharmacologic interventions to test specific hypotheses. With slight modifications, these methods can be used to culture other neuronal populations or to assess expression of other proteins of interest.
Introduction
The central nervous system (CNS) was once thought to be devoid of immune surveillance, referred to as “immune privileged1.” It is now clear that this privilege does not equate to the absolute absence of immune components, but rather, a specialized regulation that functions to limit the damage associated with immunopathology1. In fact, communication between the CNS and the immune system is an ongoing conversation that is necessary for healthy brain development and response to infections2,3.
Major histocompatibility complex class I (MHCI) molecules are polygenic and polymorphic transmembrane proteins best known for their function in presenting antigenic peptides to CD8+ T cells during infection4. Classical MHCI complexes consist of a transmembrane α-chain and an extracellular light chain, called β2-microglobulin. The α-chain contains a polymorphic groove that binds an antigenic peptide for presentation5. Proper expression of MHCI on the extracellular membrane requires coordinated action of molecular chaperones at the endoplasmic reticulum to ensure proper folding of the α-chain and β2-microglobulin along with the loading of a high affinity peptide ligand5. Only once MHCI complexes are assembled, are they exported from the endoplasmic reticulum to the plasma membrane6. Upon engagement of the cognate T cell receptor with the peptide-loaded MHCI complex, CD8+ T cells mediate cell killing by releasing lytic granules containing perforin and granzymes or by inducing apoptosis through binding Fas receptor on the target cell membrane7. Additionally, CD8+ T cells produce cytokines, such as gamma interferon (IFNγ) and tumor necrosis factor alpha (TNFα), which can activate antiviral mechanisms in infected cells without cytopathic effects8,9. For many neurotropic viruses, CD8+ T cells are necessary to clear the infection from the CNS10,11,12.
It was previously thought that neurons express MHCI only under conditions of damage, viral infection, or with in vitro cytokine stimulation. Recently, research has identified a role for neuronal expression of MHCI in synaptic remodeling and plasticity13,14. Although the precise mechanisms underlying synapse regulation are not well understood, data indicates that MHCI expression level is regulated by synaptic activity15,16. One hypothesis posits that neurons express paired immunoglobulin-like receptor B (PirB) presynaptically, which binds MHCI transynaptically13,17. This interaction initiates a signaling cascade by PirB that opposes pathways involved in synaptic remodeling, thus reinforcing and stabilizing the synaptic connection17,18,19. In the absence of neuronal activity, MHCI expression is decreased14, and the loss of MHCI results in defective synapse elimination and misorganized synaptic circuits20,21.
The assay described here, which was adapted from Chevalier et al.9, uses flow cytometric analysis to quantitatively assess extracellular protein expression of MHCI on primary cultures of murine hippocampal neurons. This protocol illustrates the initial techniques for microdissecting hippocampal tissue from embryonic mouse pups. It then details processes for enzymatic and mechanical dissociation of tissue into a single cell suspension and methods for maintaining the cultures in vitro. Because they do not divide, once they are in culture, neurons must be plated in a dish and density suitable for their experimental endpoint. Next, it outlines steps for inducing MHCI expression with beta interferon (IFNβ), immunolabeling for MHCI and neuronal nuclei marker NeuN, and analyzing cells by flow cytometry. Finally, it describes procedures for assessing the flow cytometry data to identify MHCI-positive neurons and quantifying the level of the MHCI expression. Also noted in this protocol are small adjustments that can be made in order to culture cortical neurons in addition to or instead of hippocampal neurons. This protocol can be easily modified to test specific hypotheses using genetic variations or pharmacological treatments.
Protocol
All procedures were performed in compliance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and according to the International Guiding Principles for Biomedical Research Involving Animals. The protocol was approved by the University of North Carolina at Charlotte Institutional Animal Care and Use Committee (Protocol #19-020).
1. Preparing for culture
NOTE: These procedures should be done under sterile conditions in a tissue culture-designated biosafety cabinet. See Table 1 for media and solutions.
- Sterilize all dissection tools by autoclaving in self-sealing sterilization bags.
- Prepare working stocks of poly-D-lysine for treating 12-well culture dishes.
- Dissolve poly-D-lysine in 1x borate buffer to a concentration of 100 µg/mL (10x concentrated). Store approximately 1 mL aliquots at -20 ˚C until needed.
- Dilute 10x concentrated poly-D-lysine in dPBS to 10 µg/mL (1x) and filter sterilize.
- Treat 12-well culture plates with 0.5 mL per well of 1x poly-D-lysine at room temperature for at least 1 h or overnight for using the next day. Ensure the volume of poly-D-lysine is enough to fully cover the bottom of the culture area.
- Just prior to the use, remove poly-D-lysine from culture plate, rinse 3x in sterile water, and keep in sterile water until ready to plate cells. Remove all traces of liquid by a thorough aspiration prior to plating cells.
- Prepare Neuron Growth Media and FACS buffer. Filter sterilize and store at 4 ˚C until use. Neuron Growth Media can be kept for approximately 2 wks; FACS buffer can be kept for approximately 4 wks.
2. Dissecting embryonic hippocampus
NOTE: This procedure can be performed on the benchtop as it requires the use of a stereo microscope for the removal of meninges and microdissection of the hippocampus. Adhere to strict aseptic technique to minimize potential contamination.
- Euthanize a timed-pregnant C57BL/6J female mouse at embryonic day 18 in a CO2 chamber.
- Spray the abdominal skin and fur with 70% EtOH, then pinch skin with tissue forceps and make small incision with surgical scissors. Incise the peritoneum to expose the viscera and uterus.
- Grasp the uterus with standard pattern forceps, lift out of cavity, and cut the connection to the mesometrium with fine scissors. Transfer uterus with embryos to 100 mm sterile plastic culture dish placed on ice.
- Using fine scissors and fine forceps, carefully cut the uterus and remove the embryonic sacs to release embryos. Transfer embryos to a new 100 mm sterile plastic culture dish containing dPBS placed on ice.
- Decapitate embryonic pups using fine scissors, and transfer heads to new 100 mm sterile plastic culture dish containing Hibernate-E medium placed on ice.
- To remove the brain, hold the head with a pair of fine forceps in one hand. Using curved Dumont #7 forceps in other hand, insert the tip of the forceps at the base of the skull and pinch the bone and overlying skin moving anteriorly along the midline without piercing the brain.
- Using curved forceps pull apart the skin and skull to expose the brain. Sweep the curved forceps under the brain from the exposed olfactory bulb to the cerebellum to lift the brain out of the skull. Transfer the brain to a new 100 mm sterile plastic culture dish containing Hibernate-E medium placed on ice. Repeat for each brain.
NOTE: All brains may be collected in a single culture dish unless needed to be separate for experiment-specific purposes. - Under a stereo dissection microscope, working with one brain at a time and two pairs of sterile Dumont #5 forceps, pinch off olfactory bulbs and pull away meninges. Thorough removal of the meninges is necessary to avoid culture contamination by other cells and to allow further dissection.
- Once meninges are properly removed, the superior side of the cortex opens laterally, which will expose the hippocampus. Using Dumont #5 forceps, pinch the hippocampus away from the attached cortex, and carefully transfer isolated hippocampus to a sterile 15 mL conical tube containing 5 mL of Hibernate-E medium on ice.
- Repeat the dissection for both brain hemispheres for each embryonic mouse pup and combine all hippocampi into a single 15 mL conical tube, unless needed separately for experiment specific purposes.
- To culture cortical neurons, the cortex may be separated from the subcortex and processed identically to hippocampal neurons.
NOTE: At this point, protocol may be paused. Store brains or dissected hippocampi in Hibernate-E medium supplemented with B27 (2%) at 4 ˚C for up to 1 month, though extended delay may compromise the number of viable cells.
- To culture cortical neurons, the cortex may be separated from the subcortex and processed identically to hippocampal neurons.
3. Dissociating and culturing hippocampal neurons
NOTE: All procedures should be performed under sterile conditions in a tissue culture designated biosafety cabinet.
- Prepare 0.5 mL of papain dissociation solution per embryo for hippocampal dissociation. The volume of dissociation solution needed will vary depending on the number of embryonic brains being dissected.
NOTE: For cortical neuron culture, prepare 1.0 mL of papain solution per embryo.- Dissolve 20 µL of papain suspension per 1 mL of the Hibernate E medium by vortexing for approximately 3 min.
- After papain is dissolved, add 1 µL of DNase I per 1 mL of the solution. Do not vortex the solution after DNase I has been added as this can deactivate the enzyme.
- Centrifuge 15 mL conical tube containing hippocampal tissue (step 2.10) at 1, 000 x g for 5 min. Remove the supernatant and add papain solution. Resuspend the tissue by inverting several times. Incubate at 37 ˚C for 30 min, inverting to resuspend the tissue every 10 min.
- If culturing cortical neurons, transfer all cortices to a new 100 mm sterile culture dish, carefully aspirate media from plate, and mince tissue with fine scissors or razor blade. Add papain solution to the culture dish and transfer cortical tissue with papain solution to 15 mL conical tube using a sterile transfer pipette. Incubate tissue and enzyme solution at 37 ˚C for 30 min, agitating every 10 min.
- During 30 min incubation, prepare 3 fire-polished glass Pasteur pipettes: 1 fully open, 1 half open, and 1 quarter open.
- After the 30 min incubation, centrifuge the 15 mL conical tube containing digested brain tissues at 125 x g for 10 min. Remove the enzyme solution and replace with an equal volume of fresh Hibernate E medium.
- Using fully open Pasteur pipette, triturate the tissue 10x. Let the tissue settle for 2 min, then transfer the supernatant cell suspension to a sterile 50 mL conical tube. Add an equal volume of Hibernate E medium back to the tissue.
- Using half open Pasteur pipette, triturate tissue 10x. Let the tissue settle for 2 min, then transfer the supernatant cell suspension to the same 50 mL conical tube as in the previous step. Add an equal volume of Hibernate E medium back to the tissue.
- Using quarter open Pasteur pipette, triturate tissue 10x. Let the tissue settle for 2 min, then transfer the supernatant cell suspension again to the same 50 mL conical as in previous steps. Discard any remaining tissue that hasn’t dissociated.
- Centrifuge the 50 mL conical tube containing the supernatant from the cell suspension at 125 x g for 5 min. If not already done, wash poly-D-lysine-coated plates with sterile water and remove all traces of liquid by thorough aspiration during centrifugation.
- After centrifugation, discard the supernatant and resuspend the cell pellet in 5 mL of Neuron Growth Media. Count live cells using trypan blue and hemocytometer.
- If culturing cortical neurons, add at least 10 mL of Neuron Growth Media for less dense cells and more accurate counting.
- Dilute cells with Neuron Growth Media for a final plating density of 5 x 105 viable cells per ml and add 1 mL of diluted cell suspension to each well of a 12 well plate. Maintain neurons at 37 ˚C with 5% CO2.
NOTE: Typically, the hippocampi from 1 embryonic mouse brain, i.e., 2 hippocampi, yield approximately 1 x 106 viable cells. This number is provided for experiment-planning purposes only and should not be considered a substitution for counting cells for each culture. - Change the growth media the day after culture by removing half of the volume of media from each well, i.e., 0.5 mL, and add an equal volume of fresh media to the side of the well to avoid disrupting the cultured cells. Complete half media changes twice weekly for the lifespan of the culture.
4. Assessing MHCI expression by flow cytometry
- Prepare treatment by diluting Interferon beta (IFNβ) or an equal volume of diluent in Neuron Growth Media. Since media change will be a half-volume change, prepare treatment media with 2x concentrated IFNβ. i.e., to treat 3 wells of a 12 well plate with 100 U/mL of IFNβ, prepare 1.5 mL with 200 U/mL IFNβ or and equal volume of IFNβ diluent for media-treated controls.
- Remove 0.5 mL from each well of neurons and add 0.5 mL of Neuron Growth Media with or without IFNβ to each well. Incubate in normal growth conditions (37 ˚C, 5% CO2) for 6-72 h, depending on experimental conditions and signal optimization.
- Following treatment time, remove all culture media and gently wash once in cold Neurobasal media lacking supplements (B27, L-glutamine, Pen-Strep).
- Add 0.5 mL of non-supplemented cold Neurobasal media containing 1 µg/mL of Fc block and 1 µg/mL of fluorescent conjugated anti-MHCI antibody to each well. Incubate at 4 ˚C protected from light for 45 min.
- Remove antibody-containing media and carefully wash once in cold dPBS. Add 0.5 mL room-temperature enzyme-free cell dissociation buffer to each well and agitate to dislodge cells. Watch for dissociation using inverted tissue culture microscope.
- Once cells are detached from culture dish, add 0.5 mL of FACS buffer to each well, triturate cells to disperse clumps, and transfer total volume of each well to individual 1.7 mL microcentrifuge tubes.
- Centrifuge at 1, 000 x g for 5 min. Remove the supernatant, resuspend the cell pellet in 100 µL of FACS buffer, and transfer total volume of each tube to individual wells of a 96 well U-bottom plate.
- Add 100 µL of fixative reagent to each well. Triturate several times to avoid cells clumping and incubate for 15 min at room temperature protected from light.
- Centrifuge at 500 x g for 5 min. Remove the supernatant and resuspend in 200 µl of FACS buffer.
- Centrifuge at 500 x g for 5 min. Remove the supernatant and resuspend in 100 µL of permeabilization reagent containing fluorescent-conjugated anti-NeuN antibody (1:100 dilution) to each well. Mix well, then incubate at room temperature protected from light with rocking for 20 min.
- After incubation, centrifuge at 500 x g for 5 min. Remove the supernatant and resuspend in 100 µL of FACS buffer. Repeat 2x.
- After the final wash, resuspend cells in 100 µL of 2% paraformaldehyde diluted in FACS buffer. Mix well to prevent cells from clumping. Store at 4 ˚C protected from light until ready to read on a flow cytometer. Read samples as soon as possible, within 1 week.
5. Quantifying and evaluating data
- Measure the compensation controls for each fluorescent dye and correct any spectral overlaps. Ideal compensation controls include cultured neurons that are unstained, stained with anti-MHCI only, and stained with anti-NeuN only.
- If possible, record 100,000 events for each sample (at least 10,000) and save as FCS files.
- To analyze data, using appropriate analysis software, set up sequential gates, as depicted in Figure 1A-C to select for NeuN-positive neurons.
- Plot SSC-A (log) vs FSC-A (linear). Draw a gate on the cellular population (P1) to eliminate cellular debris from the analysis.
- Within P1 cellular population, plot FSC-H (linear) vs FSC-A (linear). Draw a gate on single cell population.
- Within the single cell population, plot SSC-A (log) vs NeuN (log). Draw a gate on NeuN-positive population using unstained or MHCI-only stained cells as a guide.
- Plot a histogram of MHCI fluorescence with cellular events normalized to mode on y-axis. Using unstained or NeuN-only stained neurons as a guide, draw a horizontal gate to quantify the percent neurons positive for MHCI.
- Export the percent cells positive for MHCI, as well as the median fluorescence intensity (MFI) for MHCI on the NeuN-positive population for statistical evaluation and graphical drawing.
Representative Results
Using the procedure presented here, hippocampal tissue was dissected from prenatal mouse pups at the embryonic day 18. The tissue was dissociated into a single cell suspension using enzymatic and mechanical methods, then cultured in 12 well plates that were pre-treated with poly-D-lysine. After 7 days in vitro, cells were treated with 100 U/mL of IFNβ or media only for 72 h, which stimulated the expression of MHCI. Neurons were stained in situ for MHCI before being non-enzymatically dissociated into a single cell suspension. Neurons were fixed and permeabilized, then stained intracellularly for neuronal nuclei marker NeuN. Samples were assessed by flow cytometry and data were analyzed using associated software. Neurons were identified through sequential gating of the total events to exclude cellular debris and doublets (Figure 1A,B). Neurons were definitively identified by NeuN-positivity (Figure 1C). NeuN+ cells were further analyzed for MHCI-positivity by plotting cells on a histogram with the number of cells normalized to the mode on the y-axis and MHCI fluorescence on the x-axis. An MHCI+ gate was drawn at the point where positive and negative peaks diverged (Figure 1D). From this, the percent neurons positive for MHCI staining (Figure 1E) and the median fluorescence intensity (MFI; Figure 1F) were calculated. Results show that IFNβ treatment significantly upregulated the percentage of neurons positive for extracellular staining of MHCI, as well as the level of expression, as indicated by MFI. Statistical analysis and graphical representation were done using commercially available statistical software.
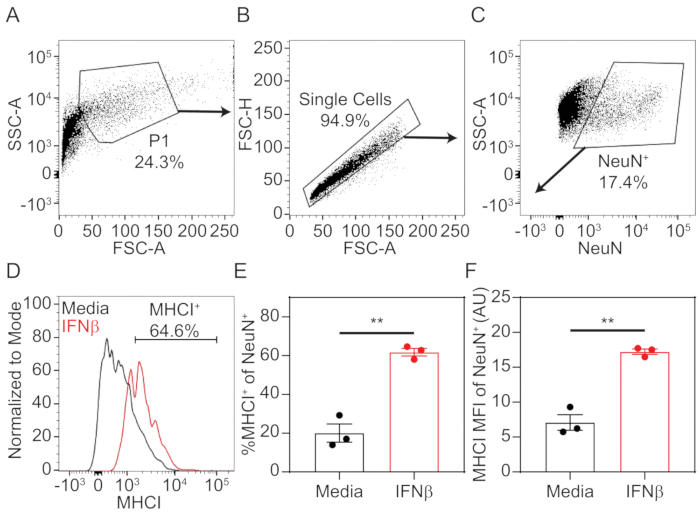
Figure 1: Representative gating strategy and MHCI quantification.
Primary hippocampal neurons were treated with 100 U/mL of IFNβ or media only. After 72 h, neurons were stained extracellularly with Pacific Blue-conjugated MHCI (1 μg/mL H2-Kb), then intracellularly labeled with PE-conjugated NeuN (1:100 dilution). Cellular fluorescence was assessed by flow cytometry, and data was analyzed. (A) Total events were plotted as SSC-A (log) vs FSC-A (linear), and cells (P1) were gated to exclude debris. (B) Within the P1 population, cells were plotted as FSC-H (linear) vs FSC-A (linear) to gate the single cell population. (C) Within the single cell population, cells were plotted SSC-A (log) vs NeuN-PE (log). NeuN+ cells were gated to identify neurons. (D) Within the neuron population, cells were plotted on a histogram with MHCI-PacBlu on the x-axis and cell numbers normalized to mode on the y-axis. A horizontal gate was drawn to quantify the percent of neurons positive for MHCI staining. (E) Quantification of percent MHCI+ of NeuN+ cells in media only and IFNβ-treated neurons. (F) Quantification of median fluorescence intensity (MFI) of MHCI on NeuN+ cells in media (black) and IFNβ-treated (red) neurons. Statistical significance was calculated by unpaired t test. **, P < 0.01. Please click here to view a larger version of this figure.
| Neuron Growth Media (50 ml) | |||
| Reagent | Final Concentration | Stock Concentration | For 50 ml |
| B27 supplement | 2% | 100% | 1.0 ml |
| L-glutamine | 2 mM | 200 mM | 0.5 ml |
| Penicillin-Streptomycin | 100 U/ml | 10,000 U/ml | 0.5 ml |
| Neurobasal media | 48.0 ml | ||
| FACS buffer (500 ml) | |||
| Reagent | Final Concentration | Stock Concentration | For 500 ml |
| Fetal Bovine Serum | 2% | 100% | 10 ml |
| EDTA | 1 mM | 500 mM | 1 ml |
| dPBS | 489 ml | ||
| Papain Dissociation Solution | |||
| Reagent | Final Concentration | Stock Concentration | For 1 ml |
| Papain Suspension | 20 U/ml | 1000 U/ml | 0.020 ml |
| DNase I | 2.5 U/ml | 2500 U/ml | 0.001 ml |
| Hibernate-E | 1.0 ml | ||
| Note: The volume of Papain Dissociation Solution needed will vary depending on the number of embryonic brains being dissected. Prepare 0.5 ml per brain for hippocampal neurons or 1.0 ml per brain for cortical neurons. | |||
| Note: Prepare dissociation solution by vortexing papain suspension in Hibernate-E for about 3 min. After papain is thoroughly dissolved, add DNase I. Do not vortex DNase I. | |||
Table 1: Media and solutions.
Discussion
This protocol describes the dissection and culture of primary hippocampal neurons from prenatal mouse pups at embryonic day 18. The use of primary neurons cultured from rodents is one of the most fundamental methodologies developed in modern neurobiology22. Although immortalized cell lines can model certain aspects of neurons, their nature as tumor-derived cells, failure to develop defined axons, and continued cell division raises doubts whether they faithfully recapitulate properties of post-mitotic neurons in vivo23. Another alternative to primary neurons is the use of human induced pluripotent stem cells (HiPSCs). The technology for using HiPSCs, especially those that are patient-derived, has advanced rapidly in recent years24. However, there are still limitations to working with HiPSCs including variability between cell lines, lack of functional maturity, and differences in epigenetic profiles25. Although there are also limitations to working with the reductionist model of primary rodent neurons, cultured neurons retain the post-mitotic nature of neurons in vivo. Also, the expansive molecular biology tools and genetic modifications available for mice favors the use of primary neurons over HiPSCs for many applications, and mouse studies can be easily translated to the more complex in vivo organism without losing the experimental genetic system. For these reasons, many researchers use primary rodent neurons to verify key aspects, if not the bulk, of their research.
For certain assays, neurons may be analyzed directly following isolation from the brain ex vivo. This is particularly desirable for experiments involving adult mice that can be subjected to specific experimental conditions or that may depend on interactions of multiple cell types; however, there are several issues that limit the type of analyses that can be done. It is technically challenging to prepare a single cell suspension of neurons from the brains of adult mice because neurons are uniquely interconnected and ensheathed by myelin26. Non-enzymatic methods of tissue trituration are inefficient at dissociating the tissue and cause cell death, whereas enzymatic preparations often cleave cell surface antigens27. Furthermore, while myelin is largely absent from embryonic mice, it comprises about 20% of the adult brain, and can impair viable cell isolation and impede flow cytometry analysis28. Many of the techniques that have been developed ultimately strip neurons of their cytosol and leave small, rounded cell bodies that consist primarily of nuclei29. Although this is acceptable for some analyses, this is not appropriate for quantifying cytoplasmic or extracellular protein expression. Furthermore, the reductionist cell culture system allows testing specific mechanistic questions on a shorter time scale than is frequently possible with an in vivo system.
Also described in this protocol are methods for stimulating MHCI expression pharmacologically with IFNβ, and the quantification of extracellular MHCI expression by flow cytometry. Stimulation by IFNβ is a useful positive control for testing other experimental conditions, but it may be noted that IFNγ and kainic acid can also stimulate MHCI expression in neurons9,30, while tetrodotoxin decreases MHCI expression14. Previous methods for detecting MHCI expression relied on in situ hybridization and immunohistochemical analysis14,15,20,31. While mRNA-based assays, such as in situ hybridization and qRT-PCR, can determine the spatiotemporal localization, cell type specificity, and levels of gene transcription, these assays cannot assess protein translation or transport to the plasma membrane. Immunohistochemical and western blot analysis can determine differences in protein expression and potentially cellular localization but can be difficult to accurately quantify. Furthermore, many MHCI antibodies recognize the complex’s tertiary structure, and are highly sensitive to conformational changes. Thus permeabilization or denaturing conditions result in loss of MHCI immunoreactivity32. The method presented here uses in situ immunostaining for MHCI, which allows for recognition of the protein by the antibody in its native conformation, followed by fixation and permeabilization methods.
With slight modifications, the methods described here can be used to culture other neuronal populations or to assess expression of other extracellular proteins of interest. Noted in this protocol are easy modifications that can be made in order to culture cortical neurons, but the methods described here may also be used to culture other neuronal populations, such as striatal neurons33. Furthermore, although this protocol specifies immunostaining of MHCI and NeuN, other cellular markers can be identified in a similar manner. In general, extracellular markers can be treated like MHCI and intracellular markers can be treated like NeuN. However, it should be noted that during the cellular dissociation step, axonal projections are severed from the soma. Because the gating strategy defined here screens out cellular debris and focuses on neuronal nuclei marker NeuN, proteins that are expressed exclusively in axonal projections may not be detected.
Until recently, neurons were thought to express MHCI only in response to damage, infection, or in vitro cytokine stimulation in order to engage cytotoxic CD8+ T cells9. New research has elucidated another function of MHCI in regulating synaptic connections during development13. The protocol described here uses IFNβ to stimulate MHCI expression in wildtype cultured neurons, but similar methods may be used with a variety of cellular stimuli or genetic modifications to test specific hypotheses. This method will enable researchers to investigate the molecular mechanisms that regulate MHCI expression, which will improve understanding of the dichotomous role of MHCI on these two distinct cellular functions.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by NIA R00 AG053412 (KEF).
Materials
| 1.5 ml Microcentrifuge Tubes | Fisher | 05-408-129 | Laboratory Supplies |
| 100 mm sterile culture dish | Fisher | FB012924 | Tissue Culture Supplies |
| 15 ml Centrifuge Tubes | Genesee | 21-103 | Laboratory Supplies |
| 50 ml centrifuge tube | Genesee | 21-108 | Laboratory Supplies |
| 500 mM EDTA | Invitrogen | AM9260G | FACS Buffer Reagent |
| 70% Ethanol | Fisher | BP82031Gal | Tissue Culture Supplies |
| 96 well U-bottom plate | Genesee | 25-221 | Flow Cytometry Supplies |
| B27 | Gibco | 17504044 | Neuron Culture Reagent |
| Borate Buffer | Thermo Scientific | 28341 | Tissue Culture Supplies |
| Borosilicate Glass Pasteur Pipette | Fisher | 13 678 20C | Laboratory Supplies |
| Cell Dissociation Buffer | Gibco | 13 151 014 | Flow Cytometry Supplies |
| DNase I | Thermo | 90083 | Dissociation Solution Reagent |
| dPBS | Gibco | 14190-235 | Tissue Culture Supplies |
| Dumont #5 Forceps | Fine Science Tools | 91150-20 | Dissection Tool |
| Dumont #7 Forceps | Fine Science Tools | 91197-00 | Dissection Tool |
| Fetal Bovine Serum | Gibco | 26140079 | Tissue Culture Supplies |
| Fine Forceps | Fine Science Tools | 91113-10 | Dissection Tool |
| Fine Scissors | Fine Science Tools | 91460-11 | Dissection Tool |
| Fix and Perm Cell Permeabilization Kit | Invitrogen | GAS003 | Flow Cytometry Supplies |
| Glutamax | Gibco | 35050061 | Neuron Culture Reagent |
| Hibernate-E Medium | Gibco | A1247601 | Neuron Culture Reagent |
| Instant Sealing Sterilization Pouches | Fisher | 181254 | Tissue Culture Supplies |
| Interferon Beta | PBL Assay Science | 12405-1 | Flow Cytometry Supplies |
| Milli-Mark Anti-NeuN-PE Antibody, clone A60 | MilliporeSigma | FCMAB317PE | Flow Cytometry Antibody |
| Neurobasal Media | Gibco | 21103049 | Neuron Culture Reagent |
| Pacific Blue anti-mouse H-2Kb Antibody | BioLegend | 116513 | Flow Cytometry Antibody |
| Papain Solution | Worthington | LS003126 | Dissociation Solution Reagent |
| Paraformaldehyde | Electron Microscopy Sciences | 15714 | Fixative |
| Penicillin-Streptomycin | Gibco | 15140122 | Neuron Culture Reagent |
| Poly-D-Lysine | Sigma | P7280 | Neuron Culture Reagent |
| Standard Pattern Forceps | Fine Science Tools | 91100-12 | Dissection Tool |
| Stereo dissection microscope | Swift | M29TZ-SM99CL-BTW1 | Dissection Tool |
| Surgical Scissors | Fine Science Tools | 91401-10 | Dissection Tool |
| Transfer Pipets | Corning | 357575 | Laboratory Supplies |
| TruStain FcX (anti-mouse CD16/32) Antibody | BioLegend | 101319 | Flow Cytometry Antibody |
| Trypan Blue | Sigma | T8154-100ML | Tissue Culture Supplies |
Referências
- Galea, I., Bechmann, I., Perry, V. H. What is immune privilege (not). Trends in Immunology. 28 (1), 12-18 (2007).
- Morimoto, K., Nakajima, K. Role of the Immune System in the Development of the Central Nervous System. Frontiers in Neuroscience. 13, 916 (2019).
- Klein, R. S., et al. Neuroinflammation During RNA Viral Infections. Annual Review of Immunology. 37, 73-95 (2019).
- Janeway, C. A. The T cell receptor as a multicomponent signalling machine: CD4/CD8 coreceptors and CD45 in T cell activation. Annual Review of Immunology. 10, 645-674 (1992).
- Peaper, D. R., Cresswell, P. Regulation of MHC class I assembly and peptide binding. Annual Review of Cell and Developmental Biology. 24, 343-368 (2008).
- Donaldson, J. G., Williams, D. B. Intracellular Assembly and Trafficking of MHC Class I Molecules. Traffic. 10 (12), 1745-1752 (2009).
- Charles, A., Janeway, J., Travers, P., Walport, M., Shlomchik, M. J. T cell-mediated cytotoxicity. Immunobiology: The Immune System in Health and Disease. 5th edition. , (2001).
- Guidotti, L. G., Chisari, F. V. Noncytolytic control of viral infections by the innate and adaptive immune response. Annual Review of Immunology. 19, 65-91 (2001).
- Chevalier, G., et al. Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS Pathogens. 7 (11), 1002393 (2011).
- Shrestha, B., Diamond, M. S. Role of CD8+ T Cells in Control of West Nile Virus Infection. Journal of Virology. 78 (15), 8312-8321 (2004).
- Plaisted, W. C., Weinger, J. G., Walsh, C. M., Lane, T. E. T cell mediated suppression of neurotropic coronavirus replication in neural precursor cells. Virology. 449, 235-243 (2014).
- Marten, N. W., Stohlman, S. A., Zhou, J., Bergmann, C. C. Kinetics of virus-specific CD8+ -T-cell expansion and trafficking following central nervous system infection. Journal of Virology. 77 (4), 2775-2778 (2003).
- Shatz, C. J. MHC class I: an unexpected role in neuronal plasticity. Neuron. 64 (1), 40-45 (2009).
- Corriveau, R. A., Huh, G. S., Shatz, C. J. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 21 (3), 505-520 (1998).
- Goddard, C. A., Butts, D. A., Shatz, C. J. Regulation of CNS synapses by neuronal MHC class I. Proceedings of the National Academy of Sciences of the United States of America. 104 (16), 6828-6833 (2007).
- Needleman, L. A., Liu, X. B., El-Sabeawy, F., Jones, E. G., McAllister, A. K. MHC class I molecules are present both pre- and postsynaptically in the visual cortex during postnatal development and in adulthood. Proceedings of the National Academy of Sciences of the United States of America. 107 (39), 16999-17004 (2010).
- Syken, J., Grandpre, T., Kanold, P. O., Shatz, C. J. PirB restricts ocular-dominance plasticity in visual cortex. Science. 313 (5794), 1795-1800 (2006).
- Atwal, J. K., et al. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science. 322 (5903), 967-970 (2008).
- Takai, T. Paired immunoglobulin-like receptors and their MHC class I recognition. Immunology. 115 (4), 433-440 (2005).
- Lee, H., et al. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature. 509 (7499), 195-200 (2014).
- Ribic, A., et al. Activity-dependent regulation of MHC class I expression in the developing primary visual cortex of the common marmoset monkey. Behavioral and Brain Functions. 7, 1 (2011).
- Sahu, M. P., Nikkilä, O., Lågas, S., Kolehmainen, S., Castrén, E. Culturing primary neurons from rat hippocampus and cortex. Neuronal Signaling. 3 (2), (2019).
- Gordon, J., Amini, S., White, M. K. General overview of neuronal cell culture. Methods in Molecular Biology. 1078, 1-8 (2013).
- Zhang, X., Hu, D., Shang, Y., Qi, X. Using induced pluripotent stem cell neuronal models to study neurodegenerative diseases. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 1866 (4), 165431 (2020).
- Berry, B. J., Smith, A. S. T., Young, J. E., Mack, D. L. Advances and Current Challenges Associated with the Use of Human Induced Pluripotent Stem Cells in Modeling Neurodegenerative Disease. Cells, Tissues, Organs. 205 (5-6), 331-349 (2018).
- Ho, H., et al. A Guide to Single-Cell Transcriptomics in Adult Rodent Brain: The Medium Spiny Neuron Transcriptome Revisited. Frontiers in Cellular Neuroscience. 12, 159 (2018).
- Panchision, D. M., et al. Optimized Flow Cytometric Analysis of Central Nervous System Tissue Reveals Novel Functional Relationships Among Cells Expressing CD133, CD15, and CD24. Stem Cells. 25 (6), 1560-1570 (2007).
- Snaidero, N., Simons, M. Myelination at a glance. Journal of Cell Science. 127 (14), 2999-3004 (2014).
- Martin, D., Xu, J., Porretta, C., Nichols, C. D. Neurocytometry: Flow Cytometric Sorting of Specific Neuronal Populations from Human and Rodent Brain. ACS Chemical Neuroscience. 8 (2), 356-367 (2017).
- Lv, D., et al. Neuronal MHC Class I Expression Is Regulated by Activity Driven Calcium Signaling. PLoS One. 10 (8), 0135223 (2015).
- Barco, A., et al. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 48 (1), 123-137 (2005).
- Glynn, M. W., et al. MHCI negatively regulates synapse density during the establishment of cortical connections. Nature Neuroscience. 14 (4), 442-451 (2011).
- Facci, L., Skaper, S. D. Culture of Rodent Cortical, Hippocampal, and Striatal Neurons. Methods Molecular Biology. 1727, 39-47 (2018).
