Using the protocol described above, we knocked down three different genes, namely, white, laccase2 (lac2), and Lens3 (Table 1), at a variety of different developmental stages of the Sunburst Diving Beetle T. marmoratus. We performed RNAi in T. marmoratus by injecting dsRNA at a very early stage during embryogenesis (Figure 1A). As some of the embryos do not survive the process and turn necrotic (Figure 1B), they need to be removed to keep the remaining embryos healthy. Exemplified here are the injections of dsRNA against the white gene. This gene is well known in Drosophila as one of three ATP-binding cassette (ABC) transporters involved in the uptake and storage of the precursors of eye pigment23. Accordingly, its loss-of-function results in an unpigmented, white-eye phenotype. Our results show that the injections of dsRNA targeting the orthologous white gene in T. marmoratus embryos leads to the loss of eye pigmentation in newly emerging larvae. In this case, wild type larvae are characterized by heavily pigmented eyes, and the RNAi knockdown of white leads to various levels of reduction and even complete elimination of eye pigment. Overall, we observed at least some reduction in eye coloration in 34% of surviving embryos (n = 35). Figure 2A compares a control individual and an individual with slightly lighter eye color. Figure 2B illustrates a more severe knockdown in an individual, in which the more ventral eyes (Eyes 2–5) of the cluster are completely unpigmented, whereas the dorsal eyes still show some pigmentation. These differences highlight how the efficiency of the knockdown can vary regionally, which is possibly related to differences in the efficacy of dsRNA penetration in dense tissue. Another individual shows essentially complete pigment loss in all eyes (Figure 2C).
To investigate how well RNAi works at the larval stage of T. marmoratus, we injected dsRNA targeting the tannic gene lac2 into second instar larvae a few days before they were due to molt into third instars (Figure 3) and evaluated the effect on the cuticular coloration of third instar larvae. Lac2 is a type of phenoloxidase that oxidatively conjugates proteins to make them insoluble, harder, and darker. Knockdown in the flour beetle T. castaneum has been shown to lead to lighter colored individuals in low doses but is considered lethal in high doses24. Figure 4 illustrates that this treatment also leads to lighter colored Sunburst Diving Beetle larvae. Specifically, in this experiment, 75% of the surviving injected larvae (n = 12) had reduced pigmentation (compared to 0% in the control group). Figure 4A shows an individual with relatively mild depigmentation, whereas Figure 4C illustrates the head of a T. marmoratus larva in which the dark coloration of the cuticle is nearly absent. Depigmentation was particularly evident for the central dark patterning that is typical for these larvae, whereas this pattern remained clearly visible in a control-injected individual (Figure 4B). In addition, a lightening of the tail trachea was observed, as depicted in Figure 4D. In the case where lac2 dsRNA was injected into third instar larvae, lighter adult individuals were obtained (Figure 5). Note that the wings of the knockdown beetle are somewhat deformed, likely due to its unusual softness.
In addition to altering morphological traits, it is possible to use RNAi to target genes that affect behavior. To demonstrate this, we performed RNAi against a key lens protein coding gene, Lens318, and injected it into second instar larvae to affect the optical properties of third instar larval eyes. Any effects on the lens observed here are likely because the eyes of T. marmoratus larvae undergo major eye growth at this transition, which also involves major optical changes of the lens25. RNAi knockdown in this experiment was highly efficient. Verification through qPCR showed a 100% success rate; out of 13 tested individuals, 12 were knocked down to less than 10% of the expression level of control individuals, with the remaining individual having an expression level of 17% of the control level (unpublished observation). At the phenotypic level, only some individuals were severely handicapped or incapable of prey capture, as is illustrated in Figure 6 for an individual that repeatedly approached its prey from a very close distance but consistently overshot it.
Table 1: Primer sequences and amplicons for white, lac2, and Lens3 proteins. Please click here to download this table.
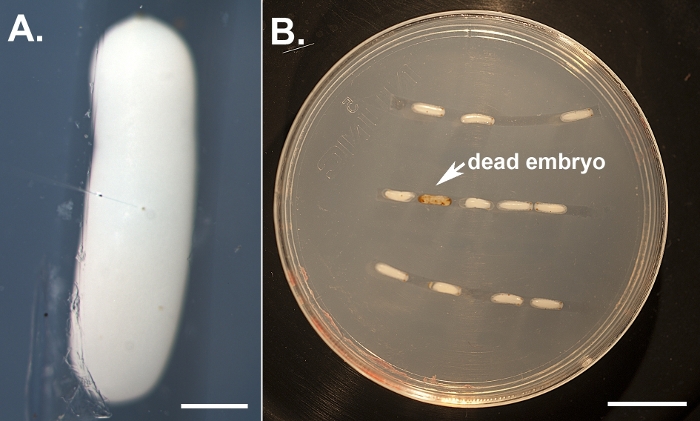
Figure 1: Illustration of embryo injections. (A) Dechorionated embryos lined up on an agarose plate and injected near their center using a microinjection needle filled with dsRNA and food-dye-containing injection buffer. The scale bar represents 500 μm. (B) An individual with a more severe knockdown. Injected embryos are kept in a humidity chamber and monitored daily to score phenotypes and remove dead individuals (which turn brown). The scale bar represents 5 mm. Please click here to view a larger version of this figure.
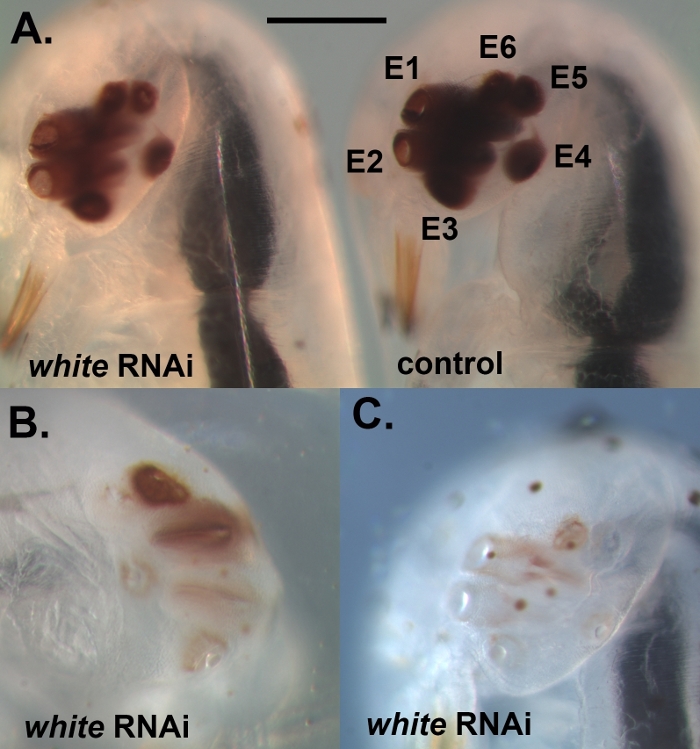
Figure 2: Example of fully developed embryos that were injected with dsRNA against white. (A) Comparison of an individual with a reduction in eye pigmentation (left) and a control-injected individual (right). E1–E6 refer to Eyes 1–6. (B) An individual with a more severe knockdown phenotype, which illustrates that, at times, some of the eyes within the cluster are more severely affected by the knockdown than others. (C) Individual with a nearly complete loss of eye pigmentation. The scale bar represents 200 μm; Panels B and C are represented at the same scale. Please click here to view a larger version of this figure.
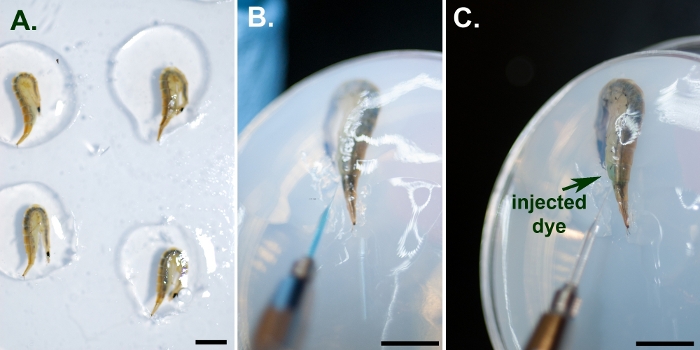
Figure 3: Illustration of larval injections. (A) Several larvae immobilized by embedding in 2% agarose with their tail spiracles left clear of any agarose. (B) Microelectrode containing the injection solution placed so that its tip can penetrate the fine membrane between two segments. (C) Blue injection dye visible at the injection site after the injection. Scale bars represent 1 cm. Please click here to view a larger version of this figure.
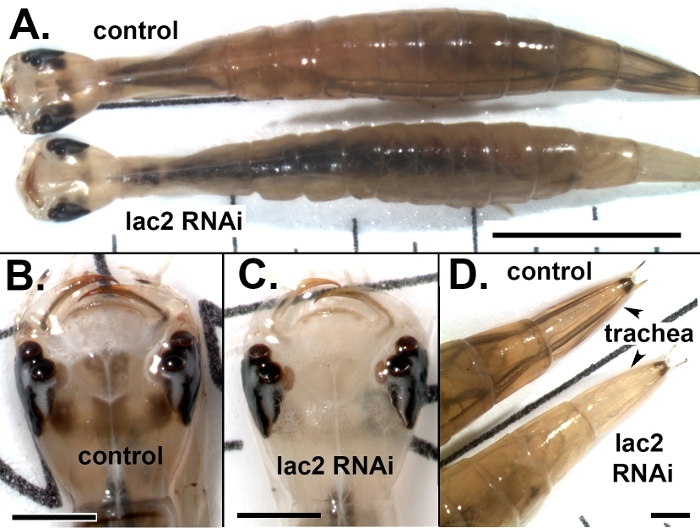
Figure 4: RNAi for laccase2 applied to second instar larvae resulting in reduced cuticle coloration in third instar larvae. (A) Relatively mild loss of coloration in a lac2 RNAi individual (bottom) when compared with a control-injected individual (top). The scale bar represents 5 mm. (B) Head of a control individual showing the characteristic dark colored pattern at the center of the head. (C) Relatively severe knockdown of lac2 leading to a nearly complete loss of central head coloration. (D) Loss of coloration in the major tail cuticle of a lac2 RNAi individual (bottom) when compared with a control-injected individual (top). Scale bars in B‒D represent 1 mm. Please click here to view a larger version of this figure.

Figure 5: RNAi for laccase2 applied to a third instar larva resulting in reduced cuticle coloration in an adult. The knockdown individual (left) is also characterized by very soft elytra when compared with the control (right). The scale bar represents 5 mm. Please click here to view a larger version of this figure.
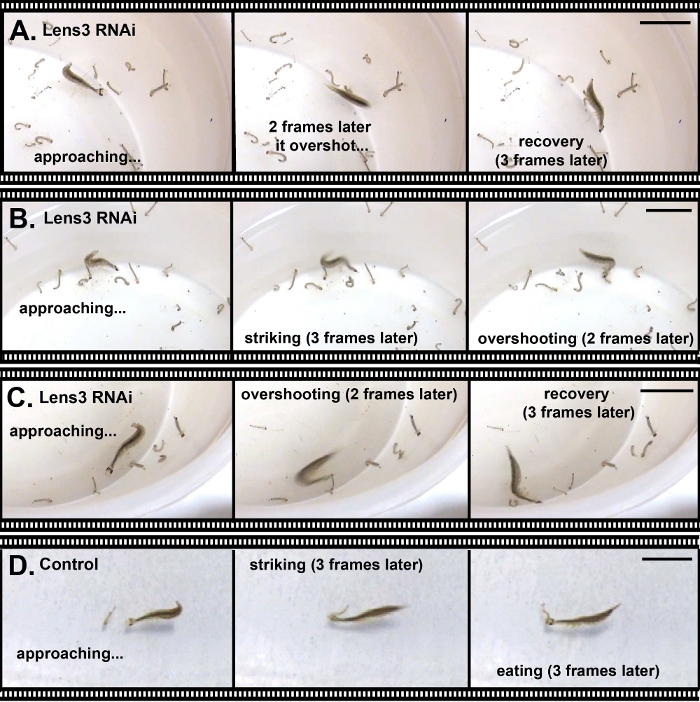
Figure 6: Knockdown of a major lens protein leading to deficiencies in prey capture. (A–C). Three examples wherein a Sunburst Diving Beetle larva, in which optical deficits were induced through RNAi, was unable to capture prey (mosquito larvae). Representative still images were selected from a video recording of characteristic prey captures. (D) Control larva catching prey. All scale bars represent 2 cm. Please click here to view a larger version of this figure.
