To demonstrate the performance of this workflow, we exploited IMAC stage tip coupled with Hp-RP stage tip fractionation to measure the phosphoproteomic changes in wild-type and snrk2-dec mutant seedlings with or without mannitol treatment for 30 minutes. Each sample was performed in biological triplicates, and the experimental workflow is represented in Figure 1. The digested peptides (400 µg) of each sample were labeled with one TMT-6plex channel, pooled and desalted. The phosphopeptides were further enriched using an IMAC stage tip, and the purified phosphopeptides were subsequently fractionated into eight fractions by a Hp-RP stage tip. Each fraction was analyzed by a 90 min LC gradient analysis. The raw files were searched using a search engine against Arabidopsis thaliana database.
A total of 11,077 unique phosphopeptides were identified corresponding to 3,630 phosphoproteins with 6,852 localized phosphorylation sites (Class I, localization probability > 0.75), indicating the wide coverage of Arabidopsis phosphoproteome. A total of 8,107 and 7,248 phosphopeptides were identified from wild-type and snrk2-dec mutant sample, respectively. This illustrates the efficiency of the workflow in providing in-depth coverage for delineating the global view of signal transduction in Arabidopsis. We compared the number of identified phosphopeptides across 8 fractions. A few phosphopeptides were identified in the first two fractions, the fraction 1 and 2. However, majority of phosphopeptides were evenly distributed in the rest 6 fractions (Figure 2A), suggesting this approach provides the capability to separate complex phosphopeptides from the plant phosphoproteome. To further demonstrate the separation efficiency of this workflow, we evaluated the overlap of phosphopeptides between two adjacent fractions (eq. F1-to-F2). Less than 5% phosphopeptides overlap in the adjacent fractions, indicating robust fractionation efficiency of the Hp-RP stage tip (Figure 2B).
Using the data obtained by the workflow, we compared the phosphoproteomic profiles of wild-type and snrk2-dec mutant upon mannitol treatment. A total of 433 and 380 phosphorylation sites were increased after mannitol treatment in wild-type and snrk2-dec mutant sample, respectively (Figure 3). Among that, 312 phosphosites showed induction (FDR < 0.01) in wild-type, but not in snrk2-dec mutant plants. The Gene Ontology (GO) analysis revealed that the function of phosphorylation and activation of protein kinase, regulation of phosphate metabolic process, signal transduction, are significantly enriched in the SnRK2-dependent phosphoproteins. Interestingly, GO term related to root development was also enriched in SnRK2-dependent group, consistent with the phenotype of root growth retardation under osmotic stress6. We also identified 116 phosphosites up-regulated by mannitol treatment in both wild-type and snrk2-dec mutant. These phosphoproteins were independent of SnRK2, or candidates that mediate osmotic stress-triggered signaling prior to SnRK2s activation. We observed that several B4 subgroup RAFs such as RAF18 (AT1G16270), RAF24 (AT2G35050), and RAF42 (AT3G46920), were significantly up-regulated by osmotic stress. Further study revealed that RAF kinases are quickly activated by osmotic stress and required for phosphorylation and activation of SnRK2s20.
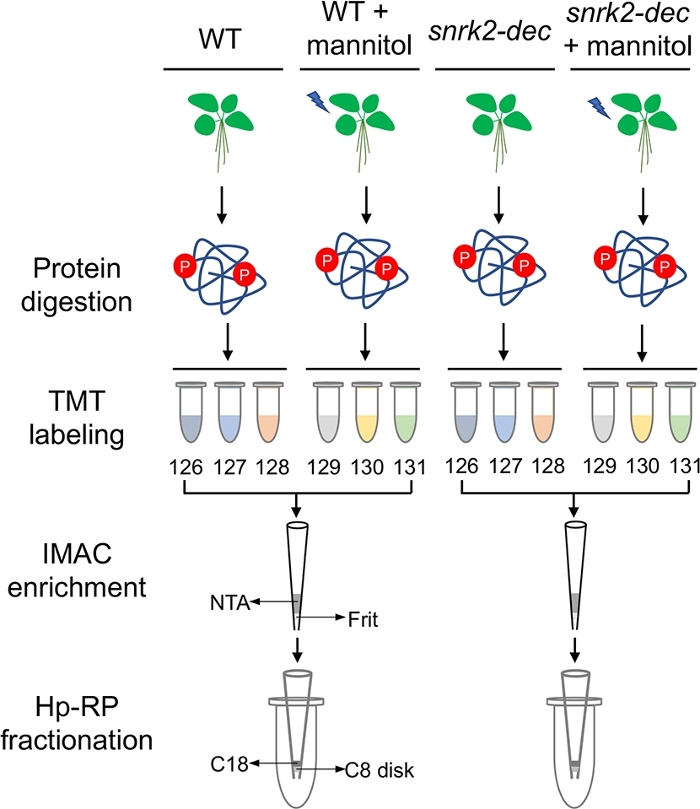
Figure 1: Workflow of stage tip-based phosphoproteomic method. Protein was extracted and digested from wild-type and snrk2-dec mutant seedlings treated with or without mannitol. The digested peptides of each replicate were labeled with a unique TMT6-plex channel. Phosphopeptides were pooled and then enriched using an IMAC stage tip. The purified phosphopeptides were separated by a Hp-RP stage tip. Each fraction was analyzed by mass spectrometer. Please click here to view a larger version of this figure.
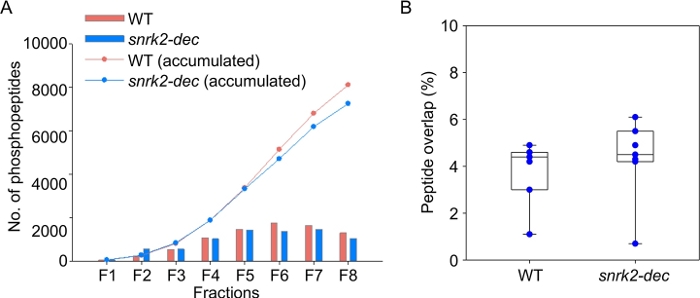
Figure 2: Phosphoproteomic profiling of wild-type and snrk2 decuple mutant seedlings by IMAC stage tip and Hp-RP fractionation. (A) The number of identified phosphopeptides per fraction. (B) The separation efficiency of the stage tip-based Hp-RP chromatography. The overlap between the adjacent fractions is represented by the percentage of the same peptide identified in the adjacent fractions. Please click here to view a larger version of this figure.
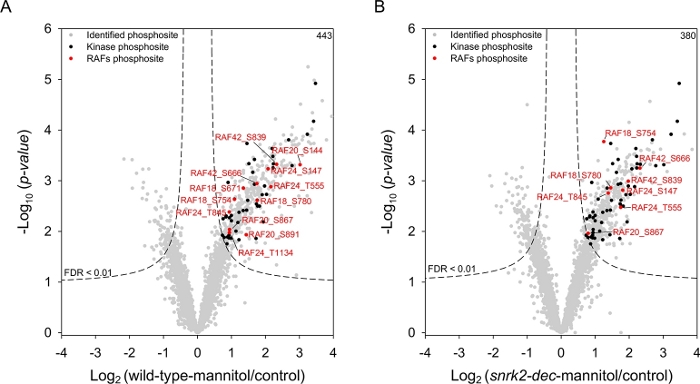
Figure 3: Quantification of the phosphoproteomic changes in response to osmotic stress. Volcano plots show the log2 fold change of phosphorylation sites in (A) wild-type and (B) snrk2-dec mutant seedlings in response to mannitol treatment. The black circle represents the kinase phosphorylation sites induced by mannitol. The red circle indicates the phosphorylation sites of RAFs up-regulated in response to mannitol. Please click here to view a larger version of this figure.
| Buffer A | 200 mM ammonium formate (NH4HCO2), pH 10.0. | |
| Buffer B | 100% ACN. | |
| Buffer 1 | 5% Buffer B, 95% Buffer A. | |
| Buffer 2 | 8% Buffer B, 92% Buffer A. | |
| Buffer 3 | 11% Buffer B, 89% Buffer A. | |
| Buffer 4 | 14% Buffer B, 86% Buffer A. | |
| Buffer 5 | 17% Buffer B, 83% Buffer A. | |
| Buffer 6 | 20% Buffer B, 80% Buffer A. | |
| Buffer 7 | 23% Buffer B, 77% Buffer A. | |
| Buffer 8 | 80% Buffer B, 20% Buffer A. | |
Table 1: Buffers for Hp-RP stage tip fractionation.
