Presented here are representative recordings from HEC slices prepared as described in this protocol. Following recovery in an interface holding chamber (Figure 1C), slices are transferred individually to a submerged recording chamber (Figure 2B). The recording chamber is supplied with carbogen-saturated ACSF using a peristaltic pump (Figure 2A). The pump first draws ACSF from a holding beaker into a heated reservoir. Carbogen lines are placed into both the holding beaker and the heated reservoir to provide continuous oxygenation of the media. A pulsation dampener, consisting of a series of air-filled syringes, is positioned in between the peristaltic pump and the recording chamber to minimize the fluctuations in flow rate produced by rapid peristalsis. The air pocket in each syringe absorbs the changes in pressure caused by each cycle of the pump, so that the recording chamber receives a smooth and consistent flow of ACSF52,53. An inline heater positioned after the pulsation dampener ensures that the temperature of the ACSF is held at 32 °C as it enters the recording chamber.
In this example, the dual-surface superfusion recording chamber consists of three 3D-printed layers (Figure 2B). The bottom layer has a rectangular cutout to fit a coverslip, secured with vacuum grease. The middle layer contains the bottom half of an elongated oval chamber, with two horizontal supports. Nylon filament is strung across these supports (roughly every 0.5 mm) and secured with cyanoacrylate adhesive. The slice will rest on top of this strung filament. The top layer contains the upper half of the oval chamber along with small wells into which the silver chloride ground pellets can be placed. The elongated oval shape of the chamber is designed to promote fast laminar flow of ACSF.
Figure 3 presents representative recordings from HEC slices prepared according to this protocol. To initially assess slice health, field postsynaptic potentials (fPSPs) are evoked in the stratum radiatum (SR) using a pipette filled with 1 M of NaCl. In healthy slices, electrical stimulation should produce a fPSP with a small presynaptic fiber volley and a large postsynaptic potential with a rapid initial descent (Figure 3B, upper left). In healthy slices, spontaneous sharp-wave ripples (SWRs) are visible as positive deflections in the LFP in the stratum pyramidale (Figure 3B, lower left). In suboptimal slices evoked fPSPs show a large fiber volley and a relatively small postsynaptic potential, and such slices do not show spontaneous SWRs (Figure 3B, right). SWRs in vitro show characteristics consistent with published descriptions: a positive field potential in the SP layer with an overlaid high frequency oscillation, paired with a negative field potential in the SR layer (Figure 3C). A single SWR recorded in CA2 is indicated with an asterisk (Figure 3C, right). SWRs in HEC slices originate within CA2/CA3 recurrent circuits and propagate to CA1. A single SWR observed in the CA2 and CA1 SP layer is indicated with an asterisk (Figure 3D, right). In this representative example, the CA2 SWR (green) leads that in CA1 (blue) by several milliseconds, as shown in the overlay of the SWR envelope (filtered at 2–30 Hz) recorded in each region.
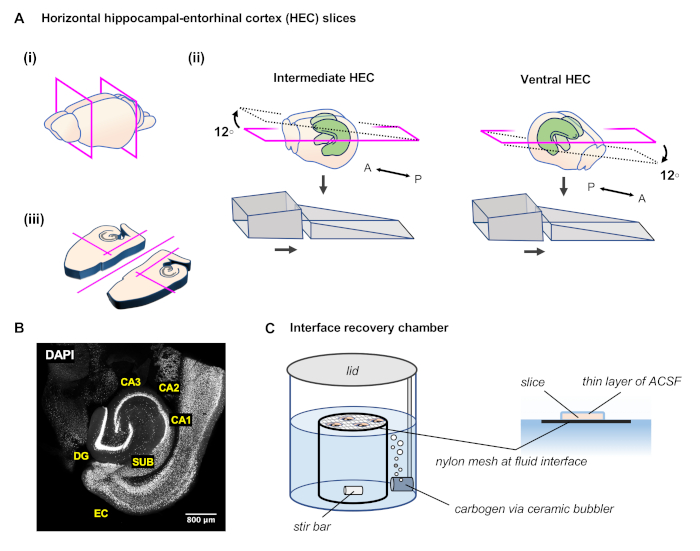
Figure 1: Preparation of horizontal angled hippocampal-entorhinal cortex (HEC) slices. (A)(i) After extracting the brain, perform two coronal cuts with a razor blade to remove the posterior and anterior portions of the brain. (ii) The agar ramp is formed of two angled portions glued to the microtome slicing platform. To prepare slices of the intermediate hippocampus, place the brain block onto the agar ramp with the anterior surface facing up the slope and making contact with the tall backing portion of the ramp. To prepare slices of more ventral hippocampus, place the brain block onto the agar ramp with the anterior surface facing down the slope, so that the posterior cut surface makes contact with the tall backing portion of the ramp. (iii) As each slice is freed, perform several more cuts with the scalpel to separate the hemispheres and remove unnecessary tissue. (B) Representative image of the resulting slice with cell nuclei labeled by DAPI. (C) In an interface recovery chamber, slices are placed on pieces of lens paper on top of a stainless steel or nylon mesh, level with the surface of the ACSF. A ceramic bubbler conveys carbogen into the chamber and a magnetic stir bar continually mixes the fluid in the chamber. A thin film of ACSF covers the top surface of the slice, enhancing diffusion of oxygen from the humid carbogen-rich air of the chamber. Please click here to view a larger version of this figure.
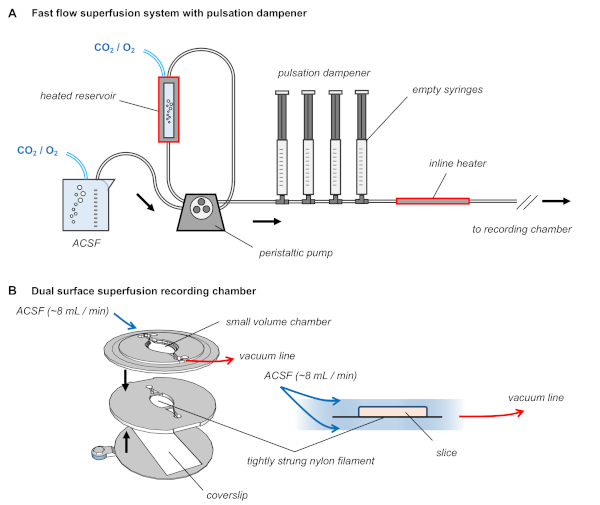
Figure 2: Dual surface superfusion recording chamber with pulsation dampener in the ACSF delivery tubing. (A) Diagram of the superfusion system. ACSF is warmed to 32 ˚C, constantly bubbled with carbogen gas, and delivered at approximately 8–10 mL/min using a peristaltic pump with a pulsation dampener consisting of a series of air-filled syringes. (B) The recording chamber consists of three 3D-printed layers, the middle of which is strung with nylon filament. The slice rests upon this strung filament and ACSF flows above and below the tissue. Please click here to view a larger version of this figure.
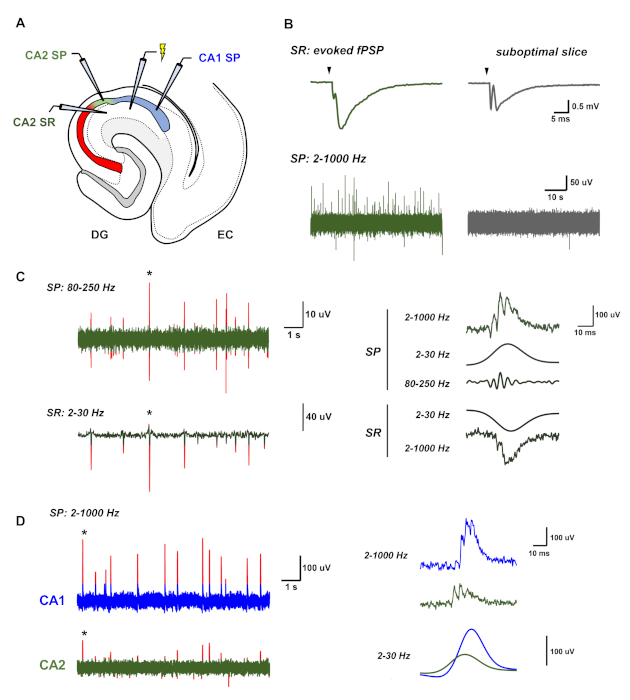
Figure 3: Representative recordings of spontaneous sharp-wave ripples from HEC slices. (A) A simplified diagram of the HEC slice showing the positions of the recording and stimulation electrodes. (B) Representative recordings of LFP activity from both an active, healthy slice and a suboptimal slice. The healthy slice (left, in green) shows large evoked field responses and spontaneous sharp-wave ripples (SWRs), visible as irregularly occurring positive deflections in the local field potential of the SP layer. In contrast, an unhealthy slice shows small evoked field responses and no spontaneous activity (right, in gray). (C) Representative recordings of SWRs in the CA2 region, consisting of a negative deflection in the LFP in the SR layer and a high frequency oscillation with an underlying positive deflection in the LFP in the SP layer. Peaks in each channel greater than three standard deviations of the signal amplitude are highlighted in red. A bandpass filter of 2–30 Hz isolates the underlying positive and negative envelope of the sharp wave in the SP and SR layer, respectively, while a bandpass filter of 80–250 Hz is used to isolate the high-frequency oscillation of the ripple in the SP layer. (D) SWRs in vitro propagate from CA2/CA3 to CA1. In these representative recordings, SWRs in CA2 (green, bottom) precede that in CA1 (blue, top) by several milliseconds. Peaks in each channel greater than three standard deviations of the signal amplitude are highlighted in red. Please click here to view a larger version of this figure.
| molecular weight (grams / mol) | final concentration (mM) | grams / 1 L sucrose cutting solution | ||
| sucrose | C12H22O11 | 342.3 | 195 | 66.749 |
| sodium chloride | NaCl | 58.44 | 10 | 0.584 |
| glucose | C6H12O6 | 180.08 | 10 | 1.801 |
| sodium bicarbonate | NaHCO3 | 84.01 | 25 | 2.1 |
| potassium chloride | KCl | 74.55 | 2.5 | 0.186 |
| sodium phosphate monobasic anhydrous | NaH2PO4 | 137.99 | 1.25 | 0.173 |
| sodium pyruvate | C3H3NaO3 | 110.04 | 2 | 0.22 |
| stock concentration (M) | final concentration (mM) | milliliters / 1L sucrose cutting solution | ||
| calcium chloride | CaCl2 | 1 | 0.5 | 0.5 |
| magnesium chloride | MgCl2 | 1 | 7 | 7 |
Table 1: Composition of sucrose cutting solution. Begin with approximately 0.75 L of purified water that has been filtered to remove trace metals and other impurities. Dissolve each solid while mixing the solution with a magnetic stir bar. Once all solids are dissolved, bubble carbogen gas through the solution for 10 min. Add the MgCl2 and CaCl2 solutions and add water to bring the total volume to 1 L. Mix with a magnetic stir bar for 10 min to ensure the solution is uniformly mixed. The osmolarity should be between 315 and 325 mOsm, and the pH should be approximately 7.4.
| molecular weight (grams / mol) | final concentration (mM) | grams / 2L ACSF | ||
| sodium chloride | NaCl | 58.44 | 125 | 14.61 |
| glucose | C6H12O6 | 180.08 | 12.5 | 4.502 |
| sodium bicarbonate | NaHCO3 | 84.01 | 25 | 4.201 |
| potassium chloride | KCl | 74.55 | 3.5 | 0.522 |
| sodium phosphate monobasic anhydrous | NaH2PO4 | 137.99 | 1.25 | 0.345 |
| ascorbic acid | C6H8O6 | 176.12 | 1 | 0.352 |
| sodium pyruvate | C3H3NaO3 | 110.04 | 3 | 0.66 |
| stock concentration (M) | final concentration (mM) | milliliters / 2L ACSF | ||
| calcium chloride | CaCl2 | 1 | 1.6 | 3.2 |
| magnesium chloride | MgCl2 | 1 | 1.2 | 2.4 |
Table 2: Composition of artificial cerebrospinal fluid. Begin with approximately 1.5 L of purified water that has been filtered to remove trace metals and other impurities. Dissolve each solid while mixing the solution with a magnetic stir bar. Once all solids are dissolved, bubble carbogen gas through the solution for 10 min. Add the MgCl2 and CaCl2 solutions and add water to bring the total volume to 2 L. Mix with a magnetic stir bar for 10 min to ensure the solution is uniformly mixed. The osmolarity should be between 315 and 325 mOsm, and the pH should be approximately 7.4.
