Preparing chromatin is a crucial step in achieving a successful ChIP. In order to prepare good quality chromatin from frozen specimens, we should ensure efficient tissue disruption before fixation to avoid the presence of tissue clumps that could hinder efficient shearing. Figure 1 shows a summarized pipeline of the protocol. Pulverization alone is not sufficient to completely dissociate the tissue since it produces cell clusters of variable size and few single cells (Figure 1a). Associating the first pulverization step with Dounce homogenization, the amount of tissue-clumps is strongly reduced and the remaining ones are smaller (Figure 1b). After the fixation and lysis steps, the number of visible single nuclei (Figure 1c) increases, while the typical spherical appearance is lost. After sonication for 28 cycles, the nuclear staining (Hoechst 33258/DAPI) is mostly not visible anymore. This is indeed a sign of successful shearing (Figure 1d). After de-crosslinking of a chromatin aliquot and visualization of the DNA on agarose gel, successful shearing can be recognized by the presence of fragments in the range of 100-300 bp. (Figure 2a) The amount of DNA can vary according to the composition of the tissue piece prepared. Such chromatin can be successfully used for ChIP-qPCR. As shown in Figure 2b the chromatin could be successfully precipitated with H3K4me3, H3K27ac (active genes related modifications) and H3K27me3 (silenced genes related modification) antibodies. Chromosome 1 Open Reading Frame 43 (C1orf43), Proteasome 20S Subunit Beta 2 (PSMB2) and Glyceraldehyde 3-phosphate dehydrogenase (mGapdh) promoter regions resulted enriched in H3K4me3 and H3K27ac in comparison with Homeobox C13 (HOXC13), Homeobox C12 (HOXC12) and the mouse Myelin Transcription Factor 1 (mMyt1) promoter regions (Table 1). This is because C1orf43, PSMB2 and mGapdh are constitutively transcribed in the liver, while HOXC13, HOXC12 and mMyt1 are silenced. H3K27me3 shows the opposite behavior confirming the success of the ChIP assay. The fact that the liver of these mice is a chimera, allowed us to analyze both murine and human chromatin. In addition, the same chromatin could be successfully used for ChIP-seq experiments. After the sequencing step, the reads were aligned to an index composed of both murine and human genomes to reduce the amount of unaligned fragments. Subsequently, the reads where separated according to species and further analyzed with EaSeq22 .The signal intensity was then measured at the transcription start site (TSS) of every gene and the result sorted for H3K4me3 signal intensity. Figure 3a and Figure 3c show both a marked presence of H3K4me3 and H3K27ac at the TSS for a considerable portion of the genes within both mouse and human chromatin. In addition to that, H3K27me3 anticorrelates with H3K4me3/H3K27ac. H3K27me3 is present on the entire length of the gene and not only at the TSS, as expected from this PTM. Figure 3b and Figure3d show the HOXC/HoxC cluster known for being enriched for H3K27me3 and transcriptionally inactive in both mouse and human livers. The profiling of H3K4me3 and H3K27ac shows peaks for this two PTMs while the signal intensity of H3K27me3 tends to be lower and more distributed.
Due to the complexity of chromatin preparation, over-fixation may happen, lysis or sonication time may be sub-optimal, big cell clumps may persist, or inadequate handling of the sample could be inadequate. These are all events affecting the quality of the preparation. In some cases, the enrichment of chromatin fragments within the correct size will still be present or will be shifted to a higher size. In other cases, there may be a loss of material due to premature lysis or unsuccessful shearing. Figure 4 shows some examples of such negative and suboptimal results. Lane 3 and 4 show an enrichment of the fragment size between 200 bp and 800 bp. However, it is clear that the fragment size spans from 100 bp to >10,000 bp. In lane 5 and 6 an enrichment in the 100-250 bp range is present with a clear loss of material during the preparation. This could explain why the sonication produced smaller fragments. Lane 7 shows a slightly sub-optimal preparation with the fragment range increased, while lane 8 shows an almost complete loss of material. This can be caused by premature nuclear lysis or insufficient tissue dissociation with consequent loss after step 5.5.
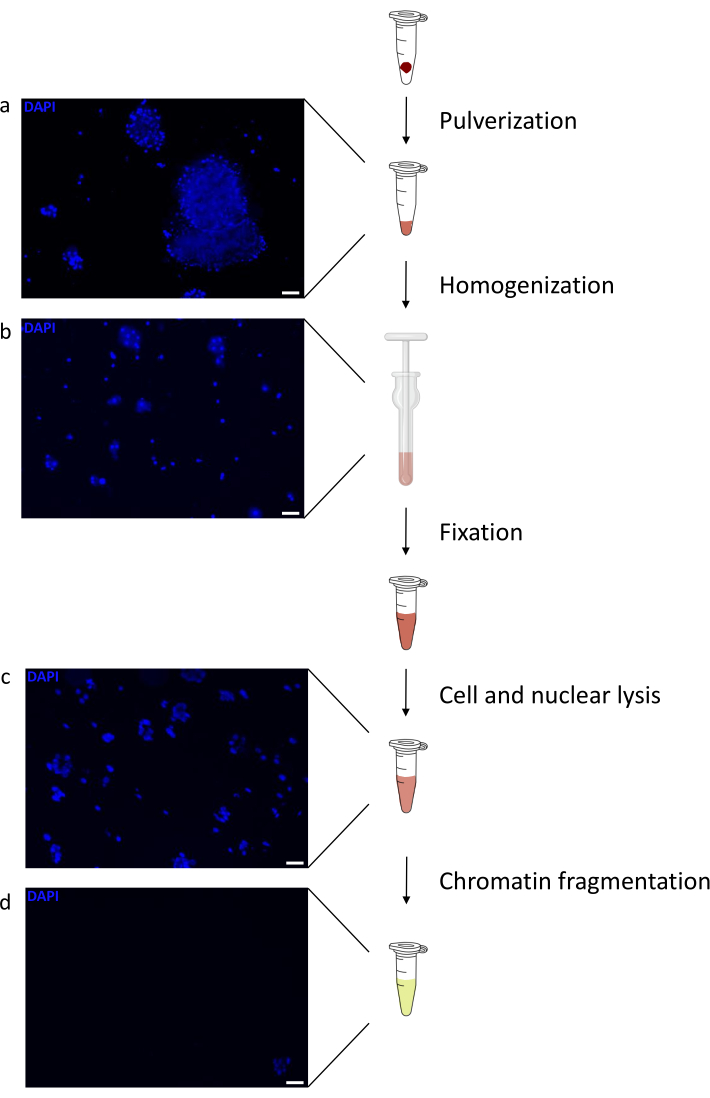
Figure 1: Chromatin preparation protocol overview. Pictures were taken after tissue pulverization (a), additional manual homogenization (b), after nuclear lysis (c) and after sonication (before centrifugation) (d). Nuclear staining was performed with Hoechst 33258/DAPI. Scale bar = 200 µm. Please click here to view a larger version of this figure.
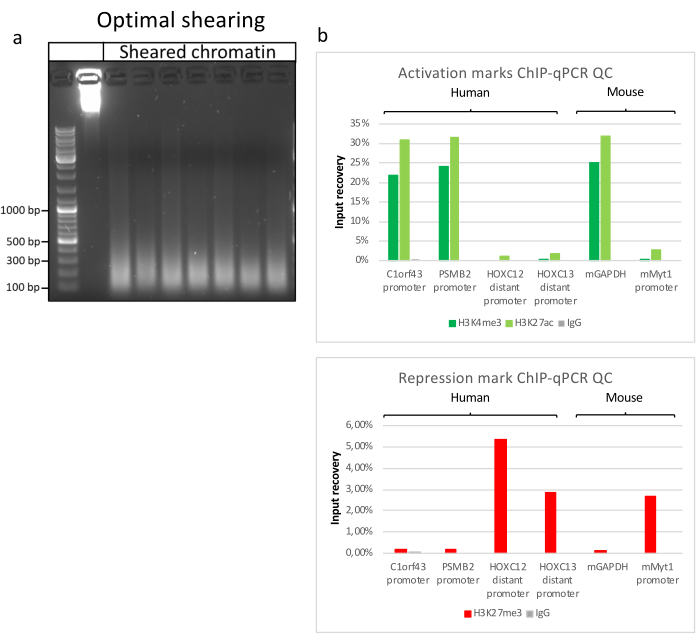
Figure 2: Representative chromatin shearing, and its quality assessed by ChIP-qPCR. 1% agarose gel with fragmented chromatin samples according to protocols from different chromatin preparations. A control of unsheared chromatin is added to ensure no chromatin/DNA degradation beforehand (a). Sheared chromatin has been tested for quality performing a ChIP-qPCR assay. H3K4me3, H3K27ac and H3K27me3 antibodies were used to precipitate the freshly prepared chromatin. (b) qPCR analysis was performed on human (C1orf43 and PSMB2), murine (Gapdh) active promoters and human (HOXC13, HOXC12), murine (Myt1) inactive promoters. Please click here to view a larger version of this figure.
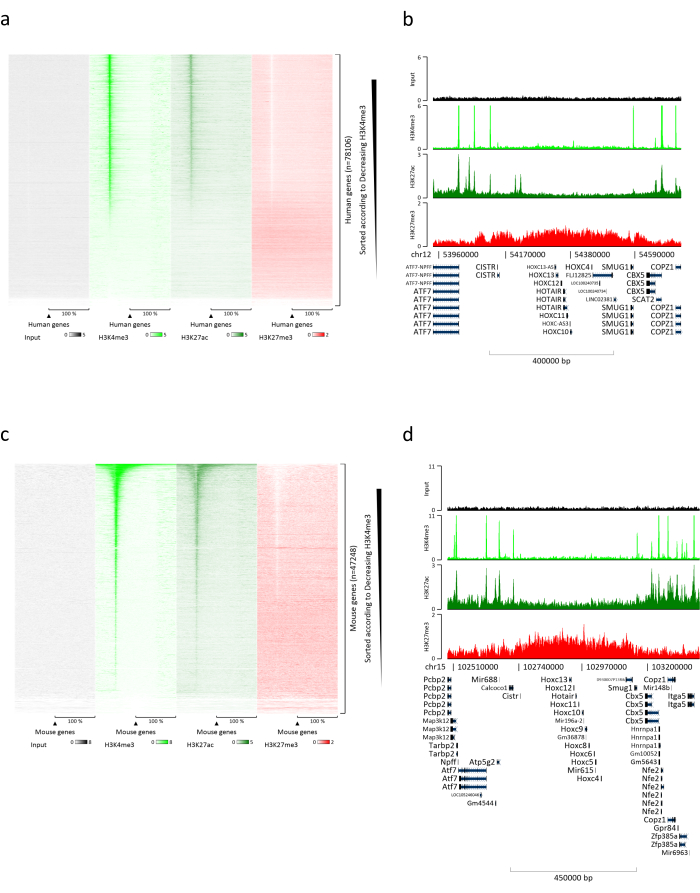
Figure 3: Representative ChIP-seq analysis. Reads have been aligned to an index created with both human and mouse genomes (hg19 and mm10). After alignment human and murine reads were separated and further analyzed. Heatmap of human genes where the signal was quantified at the TSS and showed in descending order for H3K4me3 intensity (a). Example of human gene cluster of suppressed genes (HOX cluster) surrounded by active genes (b). Heatmap of murine genes where the signal was quantified at the TSS and shown in descending order for H3K4me3 intensity (c). Example of a murine gene cluster of suppressed genes (Hox cluster) surrounded by active genes (d). All the data shown has been normalized by EaSeq per million reads. Please click here to view a larger version of this figure.

Figure 4: Suboptimal and failed chromatin preparations. 1% Agarose gel with fragmented chromatin samples according to protocol. The figure contains unsheared chromatin used as a control (Lane 2), not optimal shearing (Lane 3-4), optimal shearing with clear material loss (Lane 5-6), suboptimal shearing (Lane 7) and extensive material loss (Lane 8). Please click here to view a larger version of this figure.
| Primer name | Sequence | |
| C1orf43 promoter | Forward | AGTGGGTGGAGAATGCAGAC |
| Reverse | GAGATTACCCCACCCCATTC | |
| PSMB2 promoter | Forward | CTTATTCAACCCCCGACAAA |
| Reverse | GATGAAGGACGGTGAGAGGA | |
| HOXC13 distal promoter | Forward | GAGCCCGAGATTCACTCAAC |
| Reverse | TTATGCCCAGTTTTGGGGTA | |
| HOXC12 distal promoter | Forward | AAAGCTTCCCACTGCAAAGA |
| Reverse | AAATCTGGGGGCGAACTACT | |
| mGAPDH promoter | Forward | GGTCCAAAGAGAGGGAGGAG |
| Reverse | GCCCTGCTTATCCAGTCCTA | |
| mMYT promoter | Forward | CAGCCCAATTCTAGCCACAT |
| Reverse | CCAAAGCAGGGGAGTAGGAG |
Table 1: qPCR primers list for active and inactive genes used for ChIP-qPCR assays.
