Following the protocol described above to perform knock-in experiments at the mRosa26 locus using murine RAW264.7 macrophages, we designed a targeting vector to express human ACE2, a receptor for the SARS-Cov-2 virus (Figure 2A). Using a similar design, we generated human Jurkat T cells with knock-in of the OST-tagged RASGRP1 fusion protein (Figure 2C). After transfection of three plasmids, two of which were used for expression of CRISPR/Cas9 (DsRed2; pDsR-mR26-sg1 and pDsR-mR26-sg2 for mRosa26 knock-in; pDsR-hR26-sg1 and pDsR-hR26-sg2 for the hROSA26 knock-in) and another used as a DNA template for homologous recombination (EBFP2), double positive cells expressing two fluorescent reporters were sorted into a 96-well plate. Of the RAW264.7 cells sorted using the single cell sorting mode, 0.91% were DsRed2+ EBFP2+, but 10 cells were collected into each well (Figure 2B). The Jurkat T cells had a higher transfection efficiency, and the percentage of double positive cells was 7.91% in this representative experiment, demonstrating successful knock-in of the OST-RASGRP1 fusion protein (Figure 2D).
In the next step, flow cytometry was used to screen for the candidate wells with EBFP2-positive cells. Representative histograms showed that there were obvious EBFP2-positive populations (Figure 3). It is notable that the knock-in cells did not express DsRed2 when flow cytometry was performed two weeks after cell sorting.
For discrimination of precise knock-ins and random insertions, genomic DNA from the EBFP2-positive cells was further tested by performing PCR with primers recognizing genomic sequence external to the HAs of the targeting vector and the specific region inside the expression cassette (Figure 4A and 4B). Sequence analysis of these PCR products confirmed the occurrence of HDR at the mRosa26 target site (Figure 4C). The same genotyping strategy can be applied to validate the precise insertion of the expression cassette inthe hROSA26 locus of Jurkat T cells.
To obtain a pure population of hACE2-expressing RAW264.7 cells, we further sorted the cells and validated the presence of the fluorescent reporters following expansion using wild-type cells as controls (Figure 5A). The expanded cells were EBFP2 positive, and the hACE2 protein was readily detectable in murine RAW264.7 macrophages (Figure 5B and 5C). Similarly, we validated the expression of fluorescent reporters and the OST-RASGRP1 protein in human Jurkat T cells after screening and a second round of cell sorting (Figure 6A and 6B). In addition, affinity purification of the OST-RASGRP1 protein was performed using commercially available beads. We found a higher amount of RASGRP1 protein in the total cell lysates of knock-in Jurkat cells than in those of wild-type cells; RASGRP1 knockout Jurkat cells were used as controls (Figure 6C). After purification using the OST tag, only the knock-in samples had detectable RASGRP1 (Figure 6D).
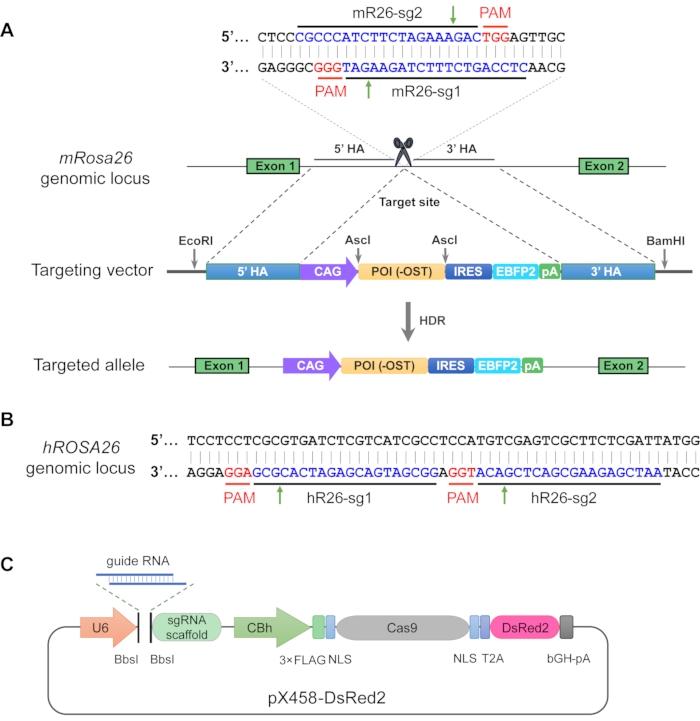
Figure 1. Gene targeting strategy for generating mRosa26/hROSA26 knock-in cell lines overexpressing a protein of interest. (A) mRosa26-specific guide RNA targeting sequences and the protospacer adjacent motifs (PAMs) in the desired insertion site are indicated in blue and red letters, respectively. Cas9 normally cleaves 3-4 bp upstream of the PAM sequence, which is indicated by green arrows. The targeting vector is used as a template for homology-directed repair (HDR) leading to the precise insertion of the expression cassette in the mouse or human genome. Each of the 1 kb sequences upstream and downstream of the desired target site are used as 5' and 3' homology arms (HAs) in the targeting vector. The HAs are separated by an expression cassette consisting of a CAG promoter, the cDNA sequence of the protein of interest (POI) with an OST-tag, an IRES-EBFP2 reporter, and a bGH-polyA signal (pA). The restriction site AscI is used for cloning of the POI, and EcoRI and BamHI are used for linearization of the targeting vector. The strategy for CRISPR/Cas9-mediated knock-in at the hROSA26 locus is similar. (B) Diagram of the human ROSA26 locus. The hR26-sg1 and hR26-sg2 targeting sequences are indicated in blue letters and the corresponding PAM sequences in red. (C) Schematic for the all-in-one CRISPR expression vector pX458-DsRed2, which contains a human U6-driven sgRNA expression cassette and Cas9-T2A-DsRed2 fluorescent reporter cassette. Two BbsI restriction sites allow for the cloning of the guide RNA. U6, human U6 RNA polymerase III promoter; sgRNA, a chimeric single-guide RNA; CBh, a chicken β-actin hybrid promoter; NLS, nuclear localization signal; T2A, Thosea asigna virus 2A self-splicing peptide; bGH-pA, bovine growth hormone polyadenylation signal. Please click here to view a larger version of this figure.
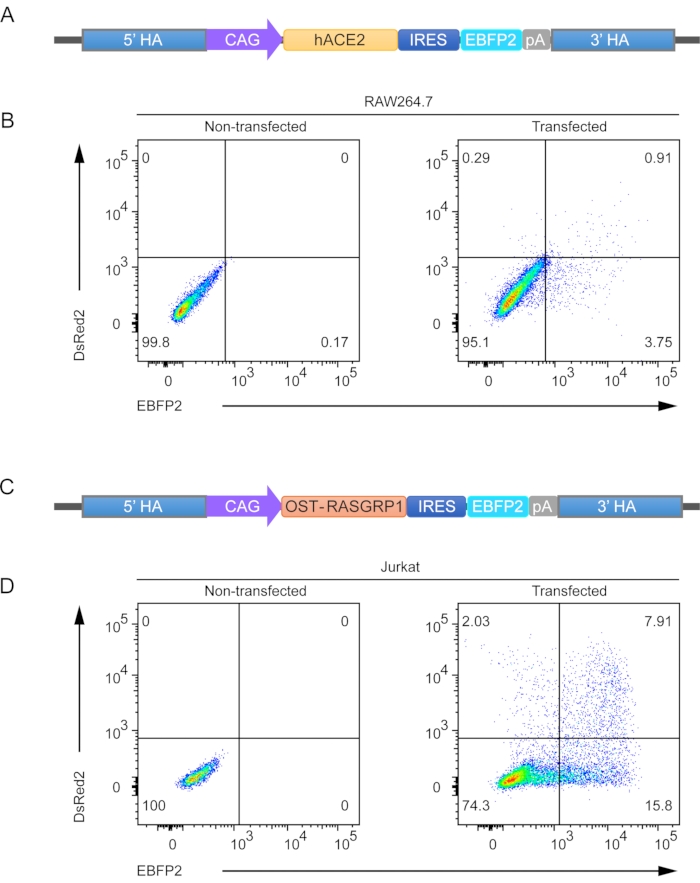
Figure 2. Single cell sorting of RAW264.7 and Jurkat cells transfected with CRISPR/Cas9 vectors and targeting vectors for homologous recombination. Targeting vectors used for stable gene expression in RAW264.7 (A) and Jurkat (C) cells. Representative flow cytometric plots of mouse RAW264.7 macrophages (B) and human Jurkat T cells (D) transfected with CRISPR/Cas9 vectors expressing the DsRed2 reporter and the targeting vector expressing the EBFP2 reporter. Cells co-expressing DsRed2 and EBFP2 were subjected to cell sorting and cultured for expansion; numbers adjacent to outlined areas indicate the percentage of cells in each gate, and non-transfected cells were used as a negative control. Please click here to view a larger version of this figure.
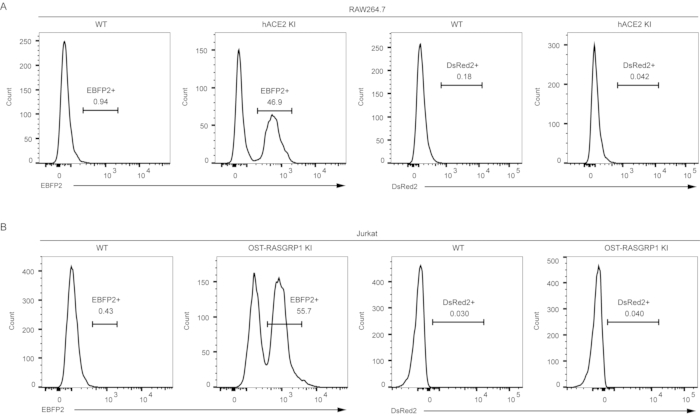
Figure 3. Screening of knock-in cells by detection of EBFP2 expression. Flow cytometric analysis of EBFP2+ and DsRed2+ RAW264.7 macrophages (A) and Jurkat T cells (B) 14 days after electroporation. In the histograms, fluorescence intensity (FI) of either EBFP2+ or DsRed2+ cells is displayed on the X-axis and the count of events in each fluorescence channel is displayed on the Y-axis. (A) The expression of EBFP2 and hACE2 was driven by the same promoter at the Rosa26 locus in RAW264.7 murine macrophages. (B) In Jurkat cells, OST-RASGRP1 expression is linked to EBFP2 expression at the human ROSA26 locus. At 14 days post-electroporation, flow cytometric analysis revealed nearly zero DsRed2+ cells among the sorted cells. Wild-type (WT) cells were used as a negative control, and KI stands for knock-in cells. Please click here to view a larger version of this figure.
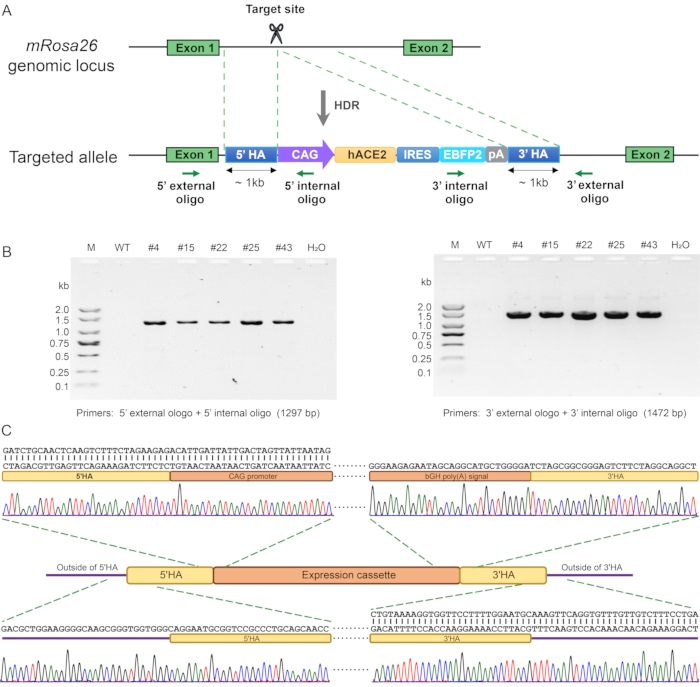
Figure 4. Screening for candidate knock-in cells by PCR and sequencing. (A) Strategy for generating hACE2 knock-in cells. The positions of PCR primers used to distinguish precise HDR and random insertion are indicated by green arrows. (B) PCR genotyping of five candidate cells (#4, #15, #22, #25, and #43) that were identified by flow cytometry screening for EBFP2 expression as exemplified in Figure 3, showed that both the 5' junction (1472 bp) and 3' junction (1472 bp) spanning the homology arms were correct. M, DNA ladder; WT, the wild-type RAW264.7 control; H2O, negative control. (C) Sanger sequencing of the PCR products from B revealed successful knock-in of the hACE2-expression cassette into the mRosa26 locus without mutations. Please click here to view a larger version of this figure.
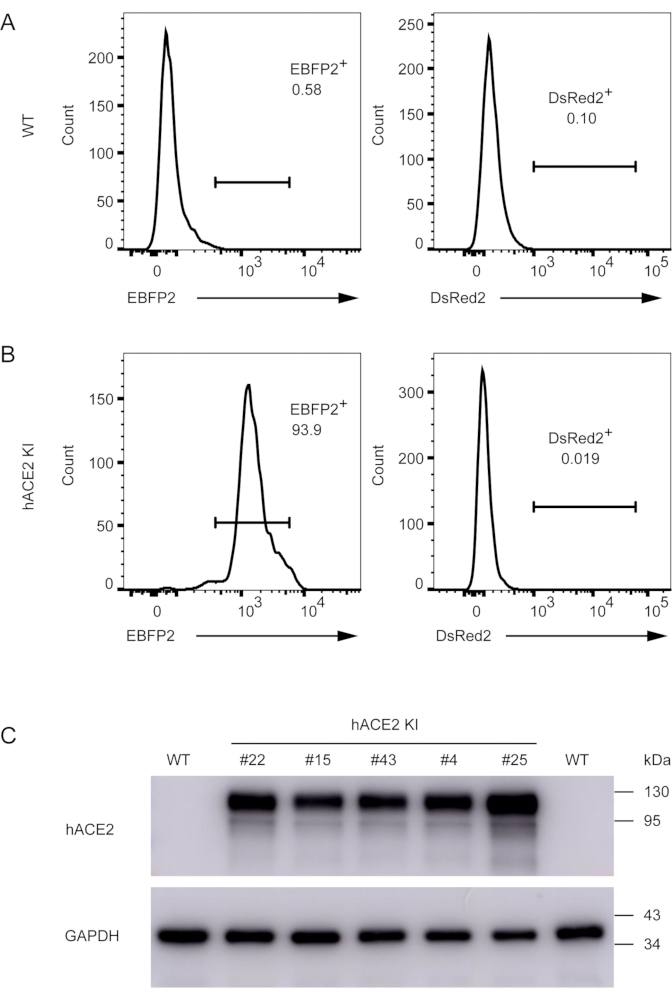
Figure 5. Validation of successful knock-in of the hACE2 expression cassette into the mRosa26 locus in RAW264.7 macrophages. (A) WT RAW264.7 macrophages were used as a negative control for flow cytometric analysis. (B) To separate the EBFP2-positive cells from the negative cells, an additional round of cell sorting was performed to obtain a population consisting of nearly 100% EBFP2+ knock-in cells, designated as hACE2 KI cells. DsRed2 expression was also examined to ensure that the CRISPR/Cas9 plasmid was not integrated into the genome of RAW264.7 macrophages. In the histograms, fluorescence intensity (FI) of either EBFP2+ or DsRed2+ cells is displayed on the X-axis and the number of events in each fluorescence channel is displayed on the Y-axis. (C) Detection of hACE2 expression by immunoblot analysis using rabbit anti-human ACE2 monoclonal antibody. Expression of hACE2 was observed in cells from multiple wells (#4, #15, #22, #25, and #43). WT RAW264.7 macrophages were used as a negative control and GAPDH was used as the loading control. Please click here to view a larger version of this figure.
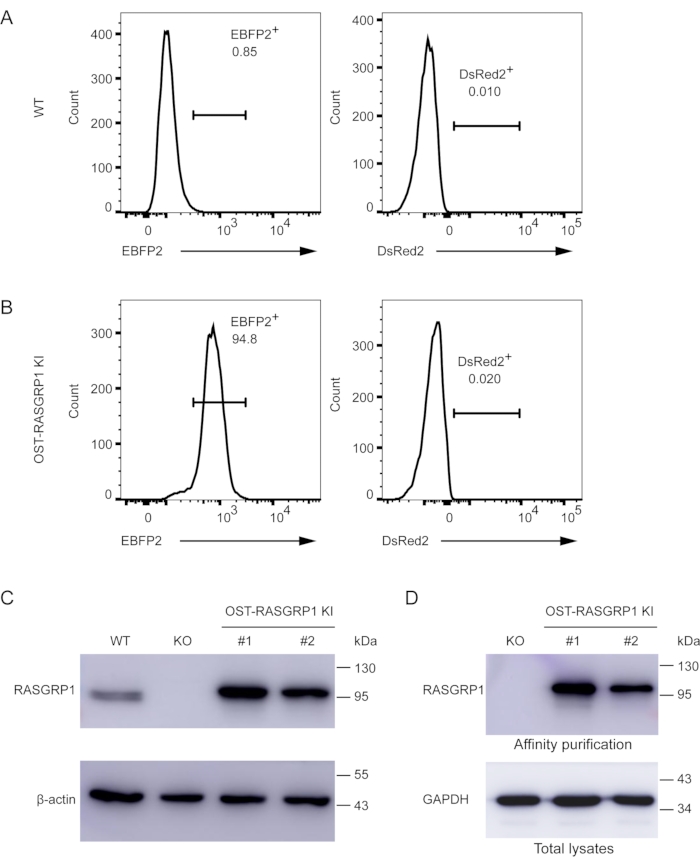
Figure 6. Validation of successful knock-in of the OST-RASGRP1 expression cassette into the hROSA26 locus in Jurkat T cells. (A) WT Jurkat T cells were used as a negative control for flow cytometric analysis. (B) EBPF2+ subpopulations of Jurkat cells were enriched by additional rounds of cell sorting and expanded; these cells were designated as OST-RASGRP1 KI cells. The knock-in cells were analyzed by flow cytometry and did not keep the DsRed2-expressing vector. In the histograms, the fluorescence intensity (FI) of either EBFP2+ or DsRed2+ cells is displayed on the X-axis and the count of events in each fluorescence channel is displayed on the Y-axis. (C) Detection of OST-RASGRP1 expression in two independent knock-in cells (#1 and #2) by immunoblot analysis using anti-RASGRP1 antibody. WT Jurkat and RASGRP1-knockout Jurkat cells were used as controls and β-actin was used as the loading control. (D) OST-mediated affinity purification was used to validate the expression of OST-RASGRP1 using RASGRP1 knockout cells as a negative control. Immunoblot analysis of equal amounts of proteins from cell lysates that were either subjected to affinity purification on Strep-Tactin Sepharose beads (Affinity purification) or directly analyzed (Total lysates) and probed with RASGRP1 or GAPDH (loading control) antibodies. Please click here to view a larger version of this figure.
Supplemental File: Supporting figures, table, and sequences Please click here to download this File.
