The Automated Crystallography Pipelines at the EMBL HTX Facility in Grenoble
Summary
Here, we describe how to use the automated macromolecular crystallography pipelines for protein-to-structure, rapid ligand-protein complex analysis and large-scale fragment screening based on the CrystalDirect technology at the HTX Laboratory in EMBL Grenoble.
Abstract
EMBL Grenoble operates the High Throughput Crystallization Laboratory (HTX Lab), a large-scale user facility offering high throughput crystallography services to users worldwide. The HTX lab has a strong focus in the development of new methods in macromolecular crystallography. Through the combination of a high throughput crystallization platform, the CrystalDirect technology for fully automated crystal mounting and cryocooling and the CRIMS software we have developed fully automated pipelines for macromolecular crystallography that can be remotely operated over the internet. These include a protein-to-structure pipeline for the determination of new structures, a pipeline for the rapid characterization of protein-ligand complexes in support of medicinal chemistry, and a large-scale, automated fragment screening pipeline enabling evaluation of libraries of over 1000 fragments. Here we describe how to access and use these resources.
Introduction
Automation has been introduced at all steps of the macromolecular crystallography experimental process, from crystallization to diffraction data collection and processing1,2,3,4,5,6,7,8,9, including a number of technologies for sample mounting10,11,12,13,14,15,16,17. This has not only accelerated the pace at which crystallographic structures are obtained but has contributed to streamline applications like structure guided drug design18,19,20,21,22,23,24. In this manuscript we describe some of the aspects of the automated crystallography pipelines available at the HTX lab in Grenoble as well as the underlying technologies.
The HTX lab at EMBL Grenoble is one of the largest academic facilities for crystallization screening in Europe. It is co-located at the European Photon and Neutron (EPN) campus togetherwith the European Synchrotron Radiation Facility (ESRF), which produces some of the world's most brilliant X-ray beams and the Institut Laue Langevin (ILL), which provides high flux neutron beams. Since the start of operations in 2003 the HTX lab has provided services to over 800 scientists and processes more than 1000 samples per year. The HTX lab has a strong focus in the development of new methods in macromolecular crystallography, including methods for sample evaluation and quality control25,26 and the CrystalDirect technology, enabling fully automated crystal mounting and processing15,16,17. The HTX lab has also developed the Crystallographic Information Management System (CRIMS), a web-based laboratory information system that provides automated communication between crystallization and synchrotron data collection facilities, enabling uninterrupted information flow over the whole sample cycle from pure protein to diffraction data. Through the combination of the capacities of the HTX facility, the CrystalDirect technology and the CRIMS software, we have developed fully automated protein-to-structure pipelines integrating crystallization screening, crystal optimization, automated crystal harvesting processing and cryocooling and X-ray data collection at multiple synchrotrons into a single and continuous workflow that can be remotely operated through a web browser. These pipelines can be applied to support rapid determination of new structures, the characterization of protein-ligand complexes and large-scale compound and fragment screening through X-ray crystallography.
The HTX lab is equipped with a nonvolume crystallization robot (including an LCP module that enables crystallization of both soluble and membrane proteins), crystal farms (at 5 °C and 20 °C), two robotic liquid handling stations to prepare crystallization screens, and two automated CrystalDirect crystal harvesters with capacity to produce and store up to 400 frozen sample pins per operation cycle. Scientists send their samples to the facility by express courier, which are then processed by dedicated technicians at the HTX lab. Scientists can remotely design crystallization screening and optimization experiments through a web interface provided by the CRIMS system. Through this interface, they can choose from a wide range of parameters and experimental protocols available at the facility to fit their specific sample requirements. Results together with all experimental parameters are made available to users in real time through CRIMS. All samples received are assayed through a specifically developed method that enables to estimate the crystallization likelihood of the sample25,26,27. Based on the results of this assay specific recommendations are made to users concerning the optimal incubation temperature and possible sample optimization experiments. Once crystallization experiments are set up, scientist can evaluate the results by looking at crystallization images collected at different time points through the web. When crystals suitable for X-ray diffraction experiments are identified, scientists can use a dedicated interface to establish a crystal mounting plan that will then be executed by the CrystalDirect robot.
The CrystalDirect technology is based on the use of a modified vapor diffusion crystallization microplate and a laser beam to mount and cryo-cool crystal samples into diffraction compatible supports closing the automation gap existing between crystallization and data collection15,16,17. Briefly, crystals are grown in a modified vapour diffusion plate, the CrystalDirect microplate. Once crystals appear the CrystalDirect harvesting robot automatically applies a laser beam to excise a film piece containing the crystal, attach it to a standard diffraction data collection pin, and cryo-cool it in a nitrogen gas stream (see Zander et al. 2016 and https://www.youtube.com/watch?v=Nk2jQ5s7Xx8 ). This technology has a number of additional advantages over manual or semi-automated crystal mounting protocols. For example, the size and shape of the crystals is not an issue, making it equally easy to harvest large crystals or microcrystals, it is often possible to avoid the use of cryo-protectants, due to the special way in which the technology operates (see reference 17, Zander et al.), making X -ray diffraction analysis much more straightforward. The laser-beam can also be used as a surgical tool to select the best parts of a sample when crystals grow on clusters or show epitaxial growth for example. The CrystalDirect technology can also be used to automated soaking experiments17. Delivery of solutions with small molecules or other the chemicals to crystals. Thereby it enables to support fully automated, large-scale compound and fragment screening. Once Crystals are harvested and cryocooled by the CrystalDirect robot, they are transferred to either SPINE or Unipuck pucks which are compatible with most synchrony macromolecular crystallography beamlines around the world. The system can harvest up to 400 pins (the capacity of the cryogenic storage Dewar) in a fully autonomous manner. CRIMS communicates with the harvester robot during the process and provides automated tracking of crystal samples (pucks and pins). Pucks are marked with both barcodes and RFID tags to facilitate sample management21,28.
CRIMS provides an application program interface (API) supporting automated communication with the ISPyB system supporting X-ray data collection management and processing at many synchrotrons in Europe and the world29. After automated crystal harvesting is completed, scientists can select crystal samples (pucks) and create sample shipments for the macromolecular crystallography beamlines at either the ESRF (Grenoble, France)7,8,9 or Petra III synchrotrons (Hamburg, Germany)18,19. CRIMS transfer the data corresponding to the selected beamline samples to the synchrotron information system along with pre-selected data collection parameters. Once the samples arrive at the selected synchrotron beamline, X-ray data collection is carried out either manually, through remote beamline operation or in a fully automated manner (i.e., at the MASSIF-1 beamline of the ESRF8 operated by the joint EMBL ESRF Joint Structural Biology Group (JSBG)). After data collection CRIMS retrieves automatically information about the results of data collection along with initial data processing results carried out by the synchrotron data processing systems and presents it to the scientist through a convenient user interface.
The HTX lab applies these automated pipelines to support three different applications, rapid determinations of new structures, rapid characterization of protein-ligand complexes and large-scale compound and fragment screening. Below we describe the how to use and operate them.
Protocol
NOTE: Funded access to these pipelines for scientists worldwide is supported through a series of funding programs. At the moment of writing this manuscript applications for access are accepted through either the iNEXT Discovery program (https://inext-discovery.eu), an European facility network to stimulate translational structural biology20 funded by the Horizon 2020 programme of the European Commission or INSTRUCT-Eric (https://instruct-eric.eu/). Contact the corresponding author for the current modalities and routes for funded access at a particular time. This protocol describes operation of the protein-to-structure pipeline and includes steps common to all our pipelines while specificities for the other two pipelines are discussed in the following section. The instructions here refer to CRIMS V4.0.
1. High Throughput Crystallization Laboratory
- Before starting, ask for registration at the HTX lab through the CRIMS system https://htxlab.embl.fr/#/. The user credentials provide remote access to all experimental design and evaluation interfaces.
- Log in to CRIMS through a web navigator (Firefox, Chrome and Safari are supported). The CRIMs web server is encrypted to prevent third parties from accessing data while it travels through the web. Once in CRIMS, a series of menus to the left of the screening help manage and create samples, request crystallization experiments, manage and visualize plates, etc. A series of video tutorials are available at https://medias01-web.embl.de/Mediasite/Showcase/embl/Channel/a2168bcaa36b4564851663e5b69594014d.
NOTE: Users sending samples by courier need to register the samples and the requested crystallization experiments through CRIMS before sending the sample to ensure they can be processed without delay upon arrival. Please, send the shipment details to htx@embl.fr.
- Log in to CRIMS through a web navigator (Firefox, Chrome and Safari are supported). The CRIMs web server is encrypted to prevent third parties from accessing data while it travels through the web. Once in CRIMS, a series of menus to the left of the screening help manage and create samples, request crystallization experiments, manage and visualize plates, etc. A series of video tutorials are available at https://medias01-web.embl.de/Mediasite/Showcase/embl/Channel/a2168bcaa36b4564851663e5b69594014d.
- Log in into CRIMS (https://htxlab.embl.fr ) with a web browser and click on the Samples Menu. This will open an interface with project and sample management tools.
- Click on the New Sample button and provide the information requested. CRIMS allows to organize samples under different projects, targets and constructs. Assign the sample to existing ones or create new ones at this point.
- Once the requested information has been entered click on Save & Make Request. Select the crystallization protocol, the crystallization screens to be used, the incubation temperature and the desired date for the experiments.
- Use the comment fields to provide indications about the samples that are important for the HTX lab operators to know. Custom screens can also be selected (see below). After submitting the crystallization request it will be validated by the HTX Lab Team and confirmation about the scheduling of the experiments will be sent via e-mail. Make sure to select experiment dates that are compatible with times required for sample shipment.
- Once the samples arrive in the facility operators at the HTX lab will carry out the experiments as requested. Once crystallization experiments are set up, confirmation will be sent via e-mail and crystallization trays will be transferred to the automated imagers. CRIMS provides access to all experimental parameters and will automatically track new imaging sessions. E-mail notifications will be automatically sent when new images are available. An thermofluor-based30 sample quality assessment experiment based on a protocol developed at the facility25,26 is carried out with every sample at this point and will be available through CRIMS.
- Images of the crystallization experiments along with the results of the sample quality assessment will be available in CRIMS shortly after the crystallization trays are set up. Click on the Thermofluor menu and navigate to the sample to see the results of the sample quality assessment experiment.
- Click on the Plates menu to see the images from the Crystallization plates. Navigate to the sample and either click on View to see the last imaging session or on the + (expand) symbol to select a different imaging session. A series of tools help to find and navigate easily through the samples. For example, clicking in a project box at the top of the screens filters samples for that project and search functions are available for most of the table columns.
- Use the plate View interface to help evaluate and score the results of the crystallization experiments. It allows navigation through the different wells of the crystallization plates, select image types (i.e., Vis, UV), select image resolution or record scores, for example. This interface also provides all experimental parameters used for the crystallization experiments including the composition of the crystallization solutions.
- Click on the Refinement menu to design crystal optimization screens based on primary hit conditions identified through the initial screening. The Chemicals and Stock Solutions submenus allow one to register and manage the crystallization stock solutions. The Screens submenu provides access to an interface to design your own optimization or custom screens.
- Select the plate type, stock solutions or gradient configurations that best fit the experimental design. It is possible to ask CRIMS to output a file directly compatible with the Formulator Robot (Formulatrix) to automatically pipette the screens into the plate or to output a printable document with the volumes for manual operation.
- Iterate through steps 1.2-1.8 to carry out crystal optimization experiments.
- Once crystals suitable for X-ray diffraction experiments are identified, navigate to the plate View interface and select the image corresponding to the right crystallization drop. Pre-stored scores will help you do this easily.
- Click either on Crystal Harvesting to record an automated crystal harvesting plan for the CrystalDirect harvester robot or on Manual Harvesting for traditional manual crystal mounting, if using CRIMS at a facility that is not equipped with CrystalDirect. Both interfaces will guide the user through the crystal harvesting process. CRIMS will automatically record and store the location of harvested crystals into either SPINE or Unipucks21.
- Select the Crystal Manager menu in CRIMS. Click on the Harvested Crystals submenu to inspect the frozen samples. When using the CrystalDirect harvester, images of the harvesting process are presented, including images of the pins with the harvested crystals.
- Select the Shipments menu to connect to either ESRF or Petra III synchrotrons and create sample shipments for X-ray diffraction analysis. Click on the Create Shipment button and select the synchrotron you want to use and the bag number (the bag password at the synchrotron is necessary here). The next series of interfaces are used to select the pukcs to be included in the shipment. The system makes it possible to provide comments to support data collection and determine data collection parameters for automated beamlines like MASSIF-1.
- If data collection is being carried out at ESRF or Petra III HTX lab, operators will transfer the samples to the beamline, data collection at other synchrotrons will be done at the user's own expense. It is possible to collect data by traveling to the synchrotron, through remote beamline operation or at MASSIF-1. In the latter case, the data collection process is fully automated. At the synchrotron, specific interfaces in ISPyB29 allow users to recover the information sent by CRIMS and associate sample pucks to it so that results of data collection are automatically tracked. For the experiments described here, data collection at synchrotrons was typically carried out with the MXcube31 software, while data processing and structure refinement was carried out with atuoPROC32, Staraninso33, BUSTER33, Pipedream32,33 and Coot35.
- Once data collection experiments have been carried out CRIMS retrieves summary information along with results of initial data processing at the synchrotron from the ISPyB29 system. Go to CRIMS Crystal Manager menu and click on the Crystal Diffraction Data sub menu. All the information and metadata regarding diffraction data collection is available. It is also possible to download processed data from the synchrotron as well as raw diffraction images. View multiple data collections or select specific datasets. Sample management tools make it possible to navigate and select samples for specific projects constructs.
NOTE: This pipeline provides fully automated operation over the internet from pure protein to X-ray diffraction results and can be operated with one or multiple samples at the same time. It can be applied to different context and project types in structural biology.
Representative Results
The automated crystallography pipeline described above has been applied to support a big number of internal and external projects with remarkable success. A few highlights include the project from Djinović-Carugo and co-workers from the Max Perutz Laboratories (Vienna) focusing on the structural and functional analysis of a dipeptidyl peptidase essential for the growth a bacterial pathogen. The rapid succession of crystallization screening, diffraction evaluation, crystal optimization and X-ray data collection cycles (up to 8 iterations for this project) enabled to obtain structural models for three different conformational states of the protein in just a few weeks, which provided key mechanistic understanding on the function of this class of proteins36 (see Figure 1).
Another example is the from Macias and co-workers from the Institute of Biomedical Research (IRB, Barcelona) that combined bioinformatics tools and structural approaches to identify new DNA binding motifs for the SMAD3 and SMAD4 transcription factors involved in cell fate regulation. This work has produced 6 high resolution structures of SMAD3 & 4 in complex with different DNA binding motifs37,38 revealing a so far unsuspected capacity of these transcription factors to recognize and bind to a diverse array of DNA sequences, which is key for the interpretation of their function in different biological contexts. These technologies have also been applied to support proprietary research in the context of drug design projects from research groups in pharma and biotech companies. For example, thanks to the rapidity contributed by these pipelines, the structural analysis of multiple ligand-target complexes can be achieved within days, which is of great value to support successive rounds medicinal chemistry optimization in the context of drug development. Finally we have also applied this infrastructure for large-scale X-ray based fragment screening39.
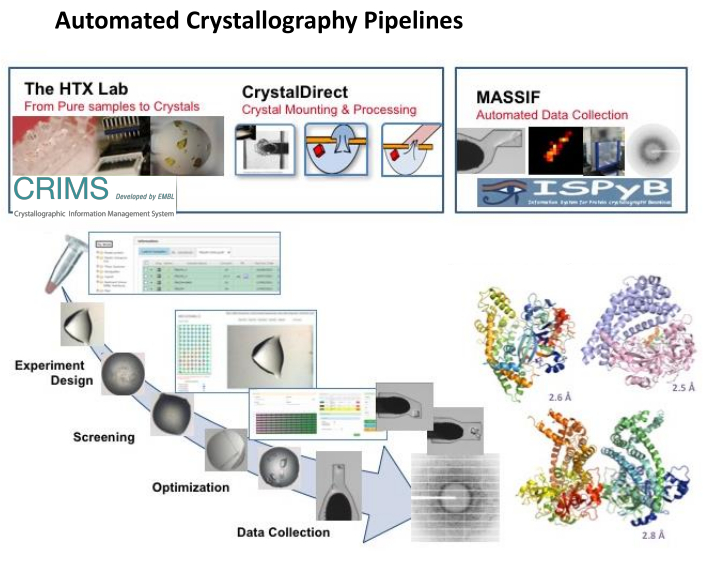
Figure 1: Automated Crystallography Pipeline. Integrated operation of the EMBL HTX lab including the CrystalDirect technology and the CRIMS software with the MASSIF-1 beamline at ESRF and automated communication between the CRIMS and ISPyB software enable to support fully automated, remote controlled protein-to-structure pipeline integrating crystallization screening and optimization, automated crystal harvesting and cryo-cooling and automated data collection and processing. The structural models correspond to three different conformational states of a protease from a pathogenic bacterium identified in a record time by applying these pipeline36. Please click here to view a larger version of this figure.
Discussion
The automated crystallography pipelines described here are available to researchers worldwide through different funding programs. Currently, funded access for crystallization experiments and the CrystalDirect technology can be obtained by applying to the iNEXT Discovery program and INSTRUCT-ERIC, while access to macromolecular crystallography beamlines at the ESRF is supported through the ESRF user access program. This approach minimizes the delay between crystal growth and measurement, accelerating the progression of very challenging projects that require diffraction-based optimization of protein production and crystallization conditions and frees scientists from complex operations associated with crystallization, crystal handling and beamline operation, rendering crystallography more accessible to non-expert groups. It can also be used for rapid exploration of crystallization additives, phasing agents or for compound screening through co-crystallization experiments. While most crystallography projects could potentially benefit from this approach, some samples may require special protocols not amenable to automation or to the pipelines presented here, for example those requiring microfluidic systems or highly specialized crystallization devices or samples that are extremely labile and would not tolerate shipment.
The CrystalDirect technology also enables automated crystal soaking17 for the characterization of of small molecule-target complexes. For this, a small aperture is created with the laser prior to the harvesting process and a drop of a solution containing the desired chemicals (i.e., phasing agents or potential ligands) is added on top, so that it enters in contact with, and diffuses into the crystallization solution eventually reaching the crystal. Chemical solutions can be formulated in water, DMSO or other organic solvents. After a certain incubation time the crystals can be harvested and analyzed by diffraction as described above. This approach has been applied to the rapid characterization of ligand-protein complexes in the context of structure-based drug design as well as to large-scale compound and fragment screening. In the latter case fragment libraries with hundreds to over a thousand fragments can be rapidly analyzed. Specific CRIMS interfaces not presented here facilitate the design and automated tracking of crystal soaking experiments, while integration between the CRIMS software and the Pipedream software suite, developed by Global Phasing Ltd (U.K) enable automated data processing, phasing, ligand identification and structure refinement over hundreds of datasets in parallel, streamlining data analysis and interpretation32,33. For example, this pipeline was recently applied to the identification of fragments binding both to the active site and several allosteric sites of Trypanosoma brucei farnesyl pyrophosphate synthase, a key enzyme of the parasite causing human African trypanosomiasis.
The pipelines presented here can contribute to accelerate the pace of discovery in structural biology and make macromolecular crystallography more accessible to a larger number of research groups. Moreover, by facilitating large-scale compound and fragment screening they can contribute to foster translational research and speed up the process of drug discovery, contributing to facilitate the development of better and safer drugs against a larger number of targets.
Declarações
The authors have nothing to disclose.
Acknowledgements
We want to thank the joint EMBL-ESRF Structural Biology Group (JSBG) for support in the use and operation of the ESRF macromolecular beamlines. We are thankful to Matthew Bowler for support with data collection at the MASSIF-1 beamline of ESRF and Thomas Schneider and the EMBL Hamburg Team for excellent support with data collection at P14 of the PetraIII synchrotron (DESY, Hamburg, Germany). The CrystalDirect harvester is developed in collaboration with the Instrumentation Team at EMBL Grenoble. This project was supported by funding from the European CommunityH2020 Programme under the projects iNEXT (Grant No 653706) and iNEXT Discovery (Grant No 871037) as well as the Région Auvergne-Rhône-Alpes through the Booster programme.
Materials
| CrystalDirect harvester | Arinax | Automated crystal mounting and cryocooling | |
| CrystalDirect Crystallization plate | Mitegen | SKU: M-XDIR-96-2 | 96-well crytsallization microplate |
| Formulator 16 | Formulatrix | For the autoamted preparation of crystallization screens | |
| Mosquito crystallization Robot | SPT Labtech | For the preparation of crystallization experiments | |
| Tecan Evo Liquid handling station | Tecan | For the preparation of crystallization solutions | |
| Spine Pucks | Mitegen | SKU: M-SP-SC3-1 | SPINE-compatible cryogenic pucks for automated synchrotron sample exchangers |
| UniPucks | Mitegen | SKU: M-CP-111-021 | Universal cryogenic pucks for automated synchrotron sample exchangers |
Referências
- Abola, E., Kuhn, P., Earnest, T., Stevens, R. C. Automation of X-ray crystallography. Nature Structural Biology. 7, 973-977 (2000).
- Banci, L., et al. First steps towards effective methods in exploiting high-throughput technologies for the determination of human protein structures of high biomedical value. Acta crystallographica. Section D, Biological. 62 (10), 1208-1217 (2006).
- Edwards, A. Large-scale structural biology of the human proteome. Annual Review in Biochemistry. 78, 541-568 (2009).
- Rupp, B., et al. The TB structural genomics consortium crystallization facility: towards automation from protein to electron density. Acta crystallographica. Section D, Biological. 58 (10), 1514-1518 (2002).
- Cipriani, F., et al. Automation of sample mounting for macromolecular crystallography. Acta crystallographica. Section D, Biological crystallography. 62 (10), 1251-1259 (2006).
- Cusack, S., et al. Small is beautiful: protein micro-crystallography. Nature Structural and Molecular Biology. 5, 634-637 (1998).
- McCarthy, A. A., et al. ID30B – a versatile beamline for macromolecular crystallography experiments at the ESRF. Journal of Synchrotron Radiation. 25 (4), 1249-1250 (2018).
- Bowler, M. W., et al. MASSIF-1: a beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. Journal of Synchrotron Radiation. 22 (6), 1540-1547 (2015).
- von Stetten, D., et al. ID30A-3 (MASSIF-3) – a beamline for macromolecular crystallography at the ESRF with a small intense beam. Journal of Synchrotron Radiation. 27 (3), 844-851 (2020).
- Viola, R., et al. First experiences with semi-autonomous robotic harvesting of protein crystals. Journal of Structural and Functional Genomics. 12, 77-82 (2011).
- Khajepour, M. Y. H., et al. REACH: Robotic Equipment for Automated Crystal Harvesting using a six-axis robot arm and a micro-gripper. Acta crystallographica. Section D, Biological crystallography. 69 (3), 381-387 (2013).
- Wagner, A., Duman, R., Stevens, B., Ward, A. . Acta crystallographica. Section D, Biological crystallography. 69, 1297-1302 (2006).
- Collins, P. M. Gentle, fast and effective crystal soaking by acoustic dispensing. Acta crystallographica. D73, 246-255 (2017).
- Deller, M. C., Rupp, B. Approaches to automated protein crystal harvesting. Acta crystallographica. F70, 133-155 (2014).
- Cipriani, F., Röwer, M., Landret, C., Zander, U., Felisaz, F., Márquez, J. A. CrystalDirect: a new method for automated crystal harvesting based on laser-induced photoablation of thin films. Acta Crystallographica Section D: Biological Crystallography. 68 (10), 1393-1399 (2012).
- Márquez, J. A., Cipriani, F. CrystalDirectTM: A Novel Approach for Automated Crystal Harvesting Based on Photoablation of Thin Films. Structural Genomics. 1091, 197-203 (2014).
- Zander, U., et al. Automated harvesting and processing of protein crystals through laser photoablation. Acta Crystallographica Section D Structural Biology. 72 (4), 454-466 (2016).
- Cianci, M., et al. P13, the EMBL macromolecular crystallography beamline at the low-emittance PETRA III ring for high- and low-energy phasing with variable beam focusing. Journal of Synchrotron Radiation. 24 (1), 323-332 (2017).
- Gati, C., et al. Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ. 1 (2), 87-94 (2014).
- iNEXT Consortium . iNEXT: a European facility network to stimulate translational structural biology. FEBS Letters. 592 (12), 1909-1917 (2018).
- Papp, G., et al. Towards a compact and precise sample holder for macromolecular crystallography. Acta Crystallographica Section D Structural Biology. 73 (10), 829-840 (2017).
- Whittle, P. J., Blundell, T. L. Protein Structure-Based drug design. Annual Review of Biophysics and Biomolecular Structure. 23, 349-375 (1994).
- Blundell, T. L., Jhoti, H., Abell, C. High-throughput crystallography for lead discovery in drug design. Nature Reviews Drug Discovery. 1, 45-54 (2002).
- Krojer, T., et al. The XChemExplorer graphical workflow tool for routine or large-scale protein–ligand structure determination. Acta Crystallographica. D73, 267-278 (2017).
- Mariaule, V., Dupeux, F., Márquez, J. A. Estimation of Crystallization Likelihood Through a Fluorimetric Thermal Stability Assay. Structural Genomics. 1091, 189-195 (2014).
- Dupeux, F., Röwer, M., Seroul, G., Blot, D., Márquez, J. A. A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Acta Crystallographica Section D Biological Crystallography. 67 (11), 915-919 (2011).
- Dimasi, N., Dupeux, F., Marquez, J. A. Expression, crystallization and X-ray data collection from microcrystals of the extracellular domain of the human inhibitory receptor expressed on myeloid cells IREM-1. Acta Crystallographica. F63, 204-208 (2007).
- Hiraki, M., Matsugaki, N., Yamada, Y., Hikita, M., Yamanaka, M., Senda, T. . RFID tag system for sample tracking at structural biology beamlines. , 060074 (2019).
- Delageniere, S., et al. ISPyB: an information management system for synchrotron macromolecular crystallography. Bioinformatics. 27 (22), 3186-3192 (2011).
- Ericsson, U., et al. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Analytical Biochemistry. 357 (2), 289-298 (2006).
- Gabadinho, J., et al. MxCuBE: a synchrotron beamline control environment customized for macromolecular crystallography experiments. Journal of Synchrotron Radiation. 17, 700-707 (2010).
- Vonrhein, C., et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallographica Section D Biological Crystallography. 67 (4), 293-302 (2011).
- Smart, O. S., et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallographica Section D Biological Crystallography. 68 (4), 368-380 (2012).
- Bezerra, G. A., et al. Bacterial protease uses distinct thermodynamic signatures for substrate recognition. Scientific Reports. 7 (1), 2848 (2017).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta Crystallographica. D66, 486-501 (2010).
- Bezerra, G. A., et al. Bacterial protease uses distinct thermodynamic signatures for substrate recognition. Scientific Reports. 7 (1), 2848 (2017).
- Martin-Malpartida, P., et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nature Communications. 8 (1), 2070 (2017).
- Aragón, E., et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-β signaling. Genes & Development. 33 (21-22), 1506-1524 (2019).
- Münzker, L., et al. Fragment‐Based Discovery of Non‐bisphosphonate Binders of Trypanosoma brucei Farnesyl Pyrophosphate Synthase. ChemBioChem. 21 (21), 3096-3111 (2020).
