The following protocol includes (1) a simplified procedure for establishing mouse hippocampal and cortical cultures based on a well-established protocol22, (2) a brief introduction to an epifluorescence microscope setup for live neurons, (3) a detailed description of loading and imaging ND6 in mouse neurons, (4) a discussion about the quantification of membrane trafficking by ND6 signal. All procedures follow the biosafety and IACUC guidelines at the Florida Atlantic University. The synthesis of ND6 has been described previously20.
1. Preparation of mouse hippocampal and cortical cultures
NOTE: If not specified otherwise, all steps must be performed in a biosafety level 2 laminar flow hood. Sterilize all tools and materials.
- Use size 5 forceps to place glass coverslips in multiwell culture plates.
NOTE: For example, a 24-well plate is ideal for 12 mm coverslips.
coverslips. - Add the appropriate volume of extracellular matrices to coat the cell culture surface (e.g., 75 μL of basement membrane matrix solution per 12 mm coverslip; see the Table of Materials)
- Place the prepared plates in the tissue culture incubator (5% CO2, 100% humidity, and 37 °C) for 1-4 h to allow basement membrane matrix crosslinking.
- Sacrifice the animals, open the skull, and transfer the whole brain to a 35 mm Petri dish with 3 mL of Hank's Balanced Salt Solution (HBSS) containing 20% fetal bovine serum (H+20). Cut the brain along the midline, and separate the cortices and hippocampi in a laminar flow dissection hood to reduce contamination.
- Use microscissors to cut the tissues into small pieces (~1 mm3). Transfer those tissues to a 15 mL conical tube containing 5 mL of H+20.
- Wash the tissues three times with 5 mL of H+20 and three times with 5 mL of HBSS.
- Add 1.5 mL of 1% trypsin with ethylenediamine tetraacetic acid and incubate at 37 °C for 10 min for enzyme digestion.
- Repeat step 1.6 and use vacuum to aspirate HBSS in the end.
- Dissociate the tissues mechanically in 1 mL of HBSS containing 2.95 g/L MgSO4•7H2O using a fire-polished glass pipette.
- Centrifuge the cell suspension at 400 × g, 4 °C for 5 min.
- Aspirate the supernatant and resuspend the cells in an appropriate volume of culture media to obtain a concentration of ~10,000,000 cells/ml.
- Plate the cells at ~1,000,000 cells/cm2 and place the culture in the incubator (5% CO2, 100% humidity, and 37 °C) for 1-4 h to facilitate cell attachment to the glass coverslips.
- Add an appropriate volume of culture medium (e.g., 1 mL per well for a 24-well plate). Add the same volume of culture medium after 1 week. After 2 weeks, replace half of the existing culture medium with fresh media every week.
2. Microscope setup for live-cell fluorescence imaging
NOTE: An exemplary imaging setup (Figure 5) includes at least an inverted fluorescence microscope (see the Table of Materials), fluorescence light source with automatic shutter, fluorescence filter sets (e.g., for imaging ND6, use 405/10 BP for excitation, 495 LP for dichroic, and 510/20 BP for emission), and a high-sensitivity camera (Table of Materials), all of which are controlled by image acquisition software (Table of Materials).
- Prepare an imaging chamber that allows temperature control and solution input/output for live-cell fluorescence imaging.
NOTE: For example, a modified open-bath imaging chamber fixed on a heating platform was used in this protocol (Figure 6). - Set up a programable device to switch the perfusion solutions and deliver the electric stimulus at defined time points during imaging.
NOTE: Hardware and software synchronization is necessary for quantitative analyses (Figure 7). For example, in this protocol, a trigger output from the imaging camera was used to start a computer program that controls an automatic switch device, which controls a multichannel perfusion system and a square-pulse stimulator. - To co-image two or more fluorescent reporters, perform multichannel fluorescence imaging either by sequentially switching filter sets or by simultaneously splitting different fluorescence emissions and projecting to the same camera.
3. Loading and imaging ND6 in neuronal cultures
- Weigh out an appropriate amount of ND6, dissolve it in dimethylsulfoxide (DMSO), and allow it to solubilize at room temperature; sonicate briefly (e.g., 3 min). Filter the crude stock solution using a 0.22 μm filter to remove large dye aggregates. After filtration, determine the dye concentration by absorption spectroscopy at 405 nm using a conventional or microvolume spectrophotometer using ε = 10,700 M-1 cm-1.
- Before application, dilute the stock solution to a concentration of 1 μM using the bath solution. Keep the stock solution at room temperature in the dark.
- Labeling of endosomes and synaptic vesicles
- To ubiquitously label endocytosed membrane compartments such as endosomes, add ND6 stock solution (e.g., 1 mM in DMSO) to the culture medium at the final concentration of 1 µM, and incubate at 5% CO2, 100% humidity, and 37 °C for 30 min. To reduce the extent of SV labeling, suppress the spontaneous neuronal activity pharmacologically. For example, use tetrodotoxin to block action potentials or 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX) and D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5) to inhibit excitatory transmission23.
- To selectively label SVs, use brief but strong stimulation to evoke presynaptic exo-/endocytosis. First, transfer the culture coverslip(s) to a 35 mm Petri dish containing 2 mL of normal Tyrode's solution at room temperature. Second, dilute the ND6 stock solution in high-K+ Tyrode's solution (90 mM KCl) at the final concentration of 1 μM. Third, replace the normal Tyrode's solution in the Petri dish with ND6-containing high-K+ Tyrode's solution and incubate at room temperature for 2 min.
- Transfer the ND6-labeled cell culture to the imaging chamber filled with prewarmed normal Tyrode's solution containing 10 μM NBQX and 10 μM D-AP5.
- Adjust the perfusion speed to ~0.2 mL/s (i.e., 1 drop per second) and start the perfusion of prewarmed normal Tyrode's solution containing NBQX and D-AP5 to remove excessive ND6 in the culture.
- Adjust the focus and locate the appropriate field of view containing healthy and well-spread neurons bearing connected neurites. Avoid areas containing unresolved dye colloids.
- Try imaging ND6-loaded cells with different exposure times to identify the best imaging settings.
NOTE: For the best exposure time, the highest pixel fluorescence intensity in the resulting image is about half of the bit range (e.g., for a 16-bit image, the bit range is from 0 to 65,535), which will allow a further increase in fluorescence when the acidic bath solution is applied. The selected exposure time should be used for all ND6 imaging. - Set up the stimulation and perfusion protocol, frame interval, and total duration. For example, in the following protocol, use a 30 s baseline, a 10 s 30 Hz electric field stimulation, 50 s recovery, 120 s 90 mM K+, 60 s recovery, 60 s Tyrode's solution with 50-mM NH4Cl, 60 s Tyrode's solution at pH 5.5, and a frame interval of 3 s.
- Start the image acquisition accompanied by the synchronized stimulation and perfusion. Monitor the simulation and solution exchange during imaging.
- Stop the perfusion after the imaging ends, remove the coverslip, and clean the imaging chamber for the next trial.
4. Quantification of membrane trafficking by a change in ND6 fluorescence
- Back up and/or make an electronic copy of all image files.
- Choose an analysis program for data extraction, e.g., an open-source image analysis software such as ImageJ24 and/or FIJI25.
- Open or import an image stack to the analysis program.
- Set the first image as the reference and align the rest to it using functions/plugins such as Rigid Registration26, which will mitigate artifacts due to xy-drifting. Refer to Figure 8 for example images.
- Average all images acquired during the 30-s pre-baseline to produce a reference image. Save a copy of this image for future reference.
- Set the intensity threshold to generate a binary image from the baseline-averaged image.
- Use Watershed in ImageJ or similar functions in another program to separate connecting neurites or cells.
- Use the Analyze Particle function with appropriate area size and circularity to solicit regions of interest (ROIs) corresponding to cell membranes, endosomes, lysosomes, or synaptic boutons. Save all selected ROIs.
- Select four background ROIs in cell-free regions within the field of view.
- Measure the average pixel intensities for each ROI in every frame of the image stack, and export the results for statistical analysis.
- Calculate the mean intensity of the background ROIs as the baseline noise, which will be subtracted from the average pixel intensities for each ROI.
- Average the three highest average intensities for every selected ROI during the application of pH 5.5 Tyrode's solution to obtain the maximal fluorescence intensity, which is defined as 100% for normalization.
- Average the three lowest average intensities for every selected ROI during the application of 50 mM NH4Cl to set the minimal fluorescence intensity, which is defined as 0% for normalization.
- Calculate the relative fluorescence changes for every ROI based on its own 0% and 100% intensities. Derive mean fluorescence changes, change kinetics, and other values/plots from individual ROI data.
SVs are specialized for neurotransmitter release via evoked exo-/endocytosis27. SVs have highly acidic lumen (i.e., pH 5.5), which is ideal for ND6. We used high K+ stimulation to evoke SV exo-/endocytosis in order to allow ND6 to access SV. Expectedly, bright green fluorescent puncta along neuronal processes showed up after loading (Figure 9A). The line profile shown in Figure 4B demonstrated a strong overlap between ND6 (green curve) and FM4-64 puncta (red curve). The strong correlation between ND6 and FM4-64 fluorescence intensities also suggest a SV-staining of ND6 (Figure 9B).
An electric stimulation and a high K+ stimulation were used to evoke the release of the readily releasable pool (RRP, i.e., SVs with high release probability) and the reserve pool SVs (i.e., SVs with low release probability), respectively. There were decreases in ND6 fluorescence in response to both stimuli (Figure 4C and Figure 4E), which suggests that ND6 resides in the SV membrane and that the SV lumen is neutralized (reported by ND6 signal decrease) during the SV release.
At the end of every trial, 50 mM NH4Cl was applied to deacidify SVs28 and pH 5.5 solution to brighten surface membrane ND6 (Figure 4D and Figure 4F). The differences in fluorescence allow us to determine ND6 in the surface membrane (~44%) and SV membrane (~56%). These number match the fractions of surface and SV membranes at the axon terminals29, suggesting that ND6 evenly distributes across membranes. Moreover, ND6 signals during two different stimuli and two pH manipulations allow us to estimate that the short electric burst mobilized about ~31% SVs and high K+ stimulation released ~70% of the remaining SVs. The rates of ND fluorescence decrease during the stimulations also match the time constant for the evoked SV exocytosis previously reported30.
ND6’s compact size leads to much less steric disturbance to cell membranes than previous tagging method14 and thus offers more accurate measurement of SV trafficking. Supporting that idea, a ten-time higher loading concentration showed no significant difference in FM4-64 destaining during stimulation (Figure 10). However, 1μM is still recommended given its moderate staining in astrocytes.
As SV exo-/endocytosis involves cholesterol (an abundant and vital lipid in the neuronal membranes), we have used ND6 imaging to ask how membrane cholesterol affects SV release and retrieval. A 1-mM for 90-min treatment of methyl-β-cyclodextrin (MβCD) can remove ~10% cholesterol from neuronal surface membrane31, mimicking aging-associated membrane cholesterol decrease. Under an exhaustive electric stimulation, the MβCD treatment significantly reduced SV release and retrieval measured by ND6 imaging (Figure 11A), which suggests that membrane cholesterol facilitates SV exo-/endocytosis32. We also evaluated cholesterol’s contribution to SV pool replenishment, which is crucial for the fidelity of neurotransmission33. Two electric stimulations with a 10-s interval was applied in the presence of Bafilomycin A1 (BafA1). BafA1 selectively inhibits v-ATPase that reacidifies SVs. By acutely blocking the reacidification of retrieved SVs, BafA1 prevents ND6 fluorescence recovery after stimulation (Figure 11B). The ND6 decrease during the second stimulus should only come from nascent SVs that replenish empty RRP. In comparison to the sham control, a significantly smaller ND6 response to the second stimulus was observed in the neurons pretreated with MβCD (i.e., smaller amplitude and faster decay of ND6 fluorescence reduction). This result supports the notion that cholesterol plays a pivotal role in recruiting new SVs to RRP.
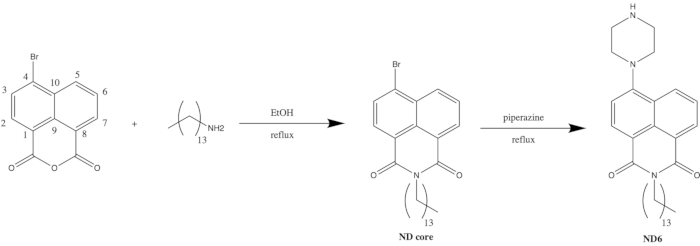
Figure 1: General synthesis scheme for ND6 probe. This figure has been modified from Thomas et al.20. Please click here to view a larger version of this figure.
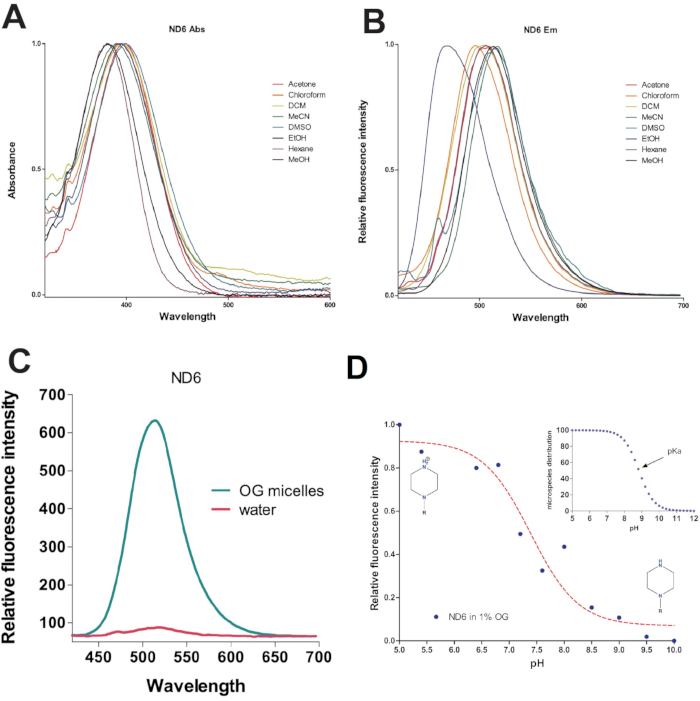
Figure 2: Properties of ND6. (A and B) Solvatochromic properties of ND6. Absorbance spectra (A) and fluorescence spectra (B) in various solvents excited at 405 nm. (C) Comparison of fluorescence intensity of ND6 in water (red) and 1% octyl glucoside solution at 1 µM. (D) ND6 fluorescence as a function of pH in 1% OG solution is proportional to protonation state of piperazine head group (calculated pKa = 7.4). Inset shows calculated protonation state of ND6 piperazine moiety (predicted pKa = 8.83). The dashed line represents fitted values. This figure has been modified from Thomas et al.20. Abbreviations: DCM = dichloromethane; MeCN = acetonitrile; DMSO = dimethylsulfoxide; EtOH = ethyl alcohol; MeOH = methyl alcohol; Abs= absorbance; Em = emission; OG = octyl glucoside. Please click here to view a larger version of this figure.
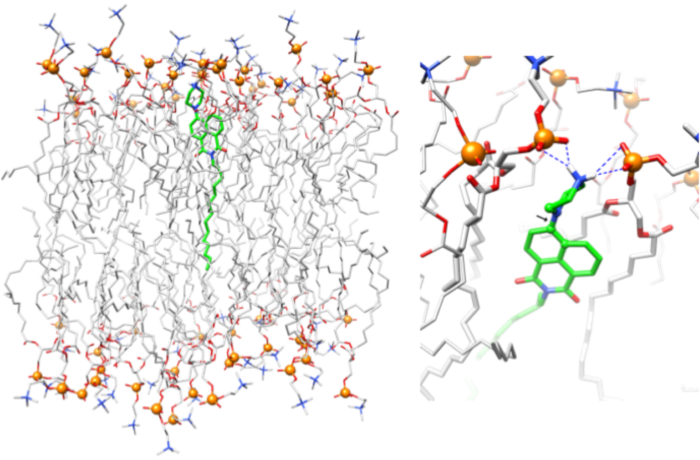
Figure 3: Snapshot from molecular dynamics simulation trajectory. Interaction of ND6 probe in POPC membrane (left panel). Piperazine head group interacts strongly with phosphate groups (right panel) through electrostatic interactions. The black arrow (right panel) points at the C-N bond between the naphthalimide ring and piperazine. Results show that the piperazine group moves only slightly with a preference for the dihedral angle (atoms showed) between 90 and 120 degrees while maintaining its chair conformation. This figure has been modified from Thomas et al.20. Please click here to view a larger version of this figure.
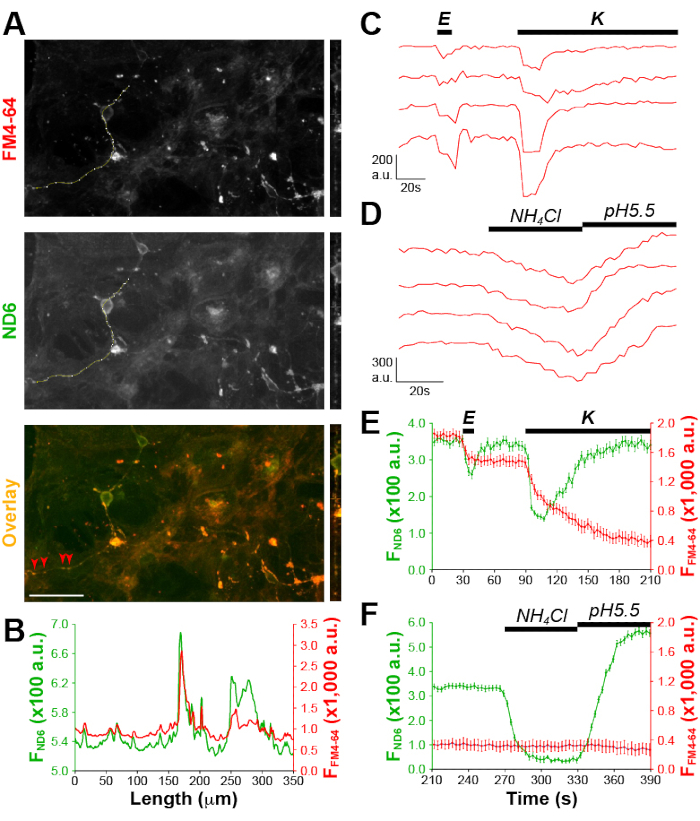
Figure 4: ND6 labels synaptic vesicles and reports their release and retrieval in the nerve terminals. (A) Sample images of FM4-64 (red), ND6 (green), and overlay (yellow). The neurites and soma of one neuron were line-profiled. The straightened line images (20-pixel width) are next to the corresponding images. Arrowheads point to synaptic boutons marked by FM4-64. Scale bars = 100 µm. (B) Line profiles of FM4-64 and ND6 fluorescence intensities exhibit significant resemblance. (C) Sample traces of ND6 fluorescence changes at synaptic boutons indicated by arrowheads in A in response to stimuli. (D) Sample traces of ND6 fluorescence changes in response to NH4Cl and pH 5.5 Tyrode's solutions. (E) The de-staining of FM4-64 is temporarily coupled to ND6 intensity changes. Data are plotted as mean ± S.E.M. (F) ND6 fluorescence intensity but not FM4-64 intensity was decreased and increased by the applications of 50 mM NH4Cl and pH 5.5 Tyrode's solutions, respectively. Data are plotted as mean ± S.E.M. This figure has been modified from Thomas et al.20. Please click here to view a larger version of this figure.

Figure 5: Imaging setup based on a Nikon-TiE inverted microscope. Annotated are four components required for live-cell fluorescence imaging. Please click here to view a larger version of this figure.

Figure 6: Stimulation-imaging setup. Setup modified from a Warner Instruments RC-26 chamber and PH-1 heating platform for temperature control and solution exchange. Please click here to view a larger version of this figure.
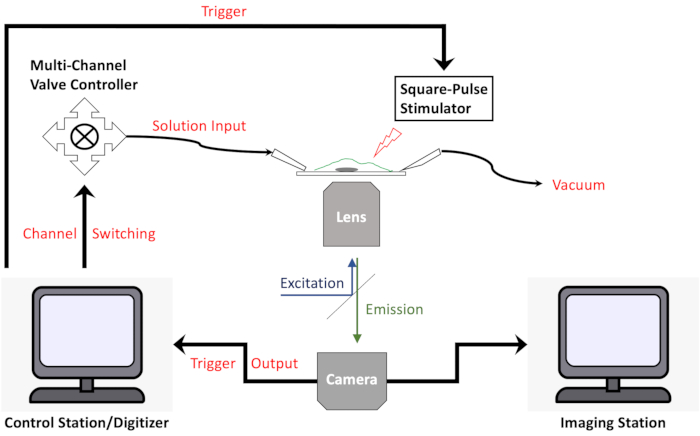
Figure 7: Diagram of device configurations for image acquisition with synchronized stimulations and solution exchanges. Please click here to view a larger version of this figure.

Figure 8: Sample images demonstrating the key steps in image analysis. Please click here to view a larger version of this figure.
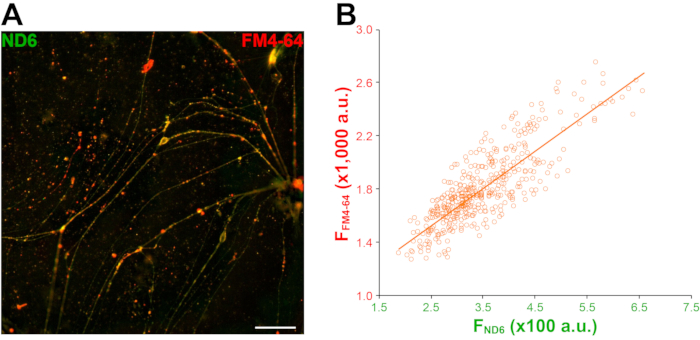
Figure 9: ND6 highlights synaptic vesicles clustered at presynaptic terminals. (A) Sample images FM4-64 (red) and ND6 (green) co-loading at high magnification. Scale bar = 30 µm. (B) Scatter plot of FM4-64 and ND6 mean fluorescence intensities at the same ROIs corresponding to synaptic boutons and linear regression fit. r = 0.8353; p = 1.7 × 10-8; fields of view N = 9; ROIs n = 450. The threshold for FM4-64 is 1,200 au (arbitrary unit) and 160 au for ND6. This figure has been modified from Thomas et al.20. Abbreviation: ROIs = regions of interest. Please click here to view a larger version of this figure.
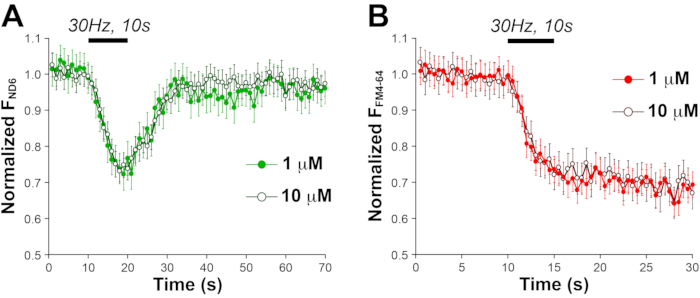
Figure 10: ND6 does not intervene with SVs. (A) ND6-represented SV turnover (by electric stimulus) after 1 or 10 µM ND6 loading. (B) FM4-64-measured SV turnover (with the electric stimulus). This figure has been modified from Thomas et al.20. Abbreviation: SVs = synaptic vesicles. Please click here to view a larger version of this figure.
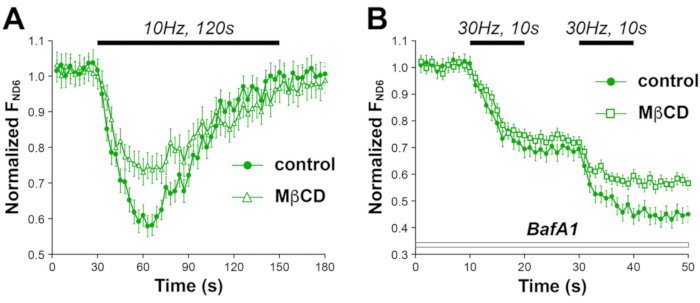
Figure 11: Cholesterol reduction impairs SV turnover. (A) ND6-represented SV turnover under 10 Hz, 120 s electric stimulus in control and MβCD-treated neurons. (B) Bafilomycin A1 prevents the reacidification of recycled SVs (i.e., no ND6 fluorescence recovery after stimulation-evoked decrease) and further elucidates MβCD's impact on SV replenishment. This figure has been modified from Thomas et al.20. Abbreviation: SV = synaptic vesicle; MβCD = methyl-β-cyclodextrin; BafA1 = Bafilomycin A1. Please click here to view a larger version of this figure.
