After calculating mean ± SEM for each group at all time points, the data is presented in line graph format with the Y-axis as the Relative Fluorescent Intensity and the X-axis as Time. Supplementary File 1 provides an example of raw data from a kinetic read of the 96-well plate in a spreadsheet format.
In this study, the optimal results presented in Figure 3A, and Table 3 demonstrate that 1) co-culture with MSC enhances the phagocytic activity of the macrophage, 2) IFN-y treatment reduces the activity of the macrophage, and 3) co-culture with MSC partially rescues MΦ phagocytic activity. Optimal cell densities are critical in these studies, and when MΦ are plated at too low of a density, changes in fluorescence intensity cannot be detected (Figure 3B). Figure 3C represents data from a study where the MΦ were plated at too high of a density and fluorescence intensity is elevated rapidly in all groups, and differences cannot be discerned. The dynamic imaging videos confirm that the fluorescent intensity changes result from phagocytosis and not acidification of the medium. They also provide qualitative data and a visual representation of the rate and extent of phagocytic activity (Figure 4).
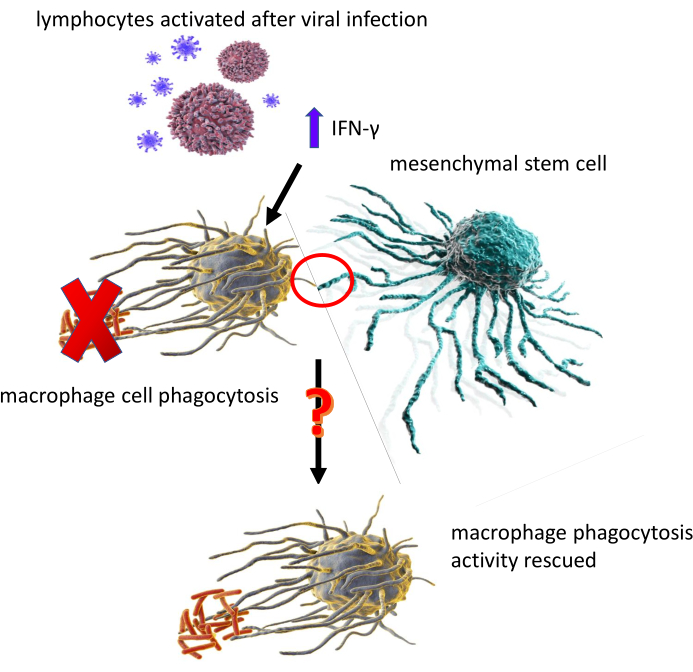
Figure 1: An illustration depicting the central question of the data presented, which is "Can MSC recover the phagocytic activity of IFN-γ treated MΦ?". Please click here to view a larger version of this figure.
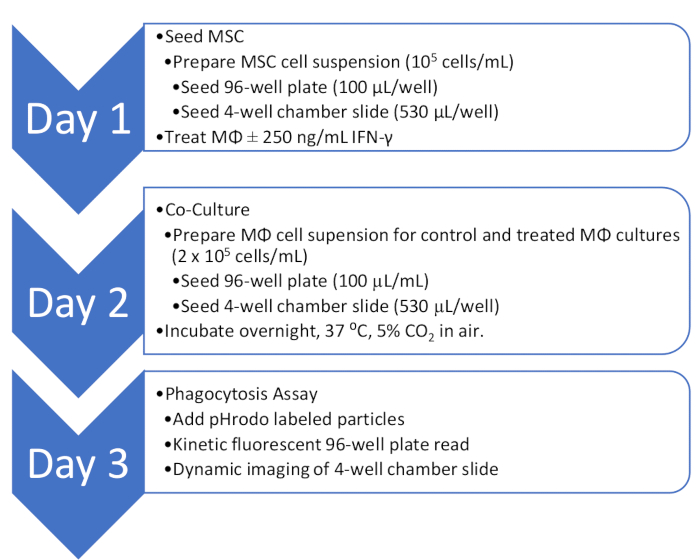
Figure 2: Overview of the co-culture and phagocytosis assay workflow. An outline of the workflow for quantitative and qualitative analysis of MΦ phagocytosis activity in co-culture with MSC. Please click here to view a larger version of this figure.
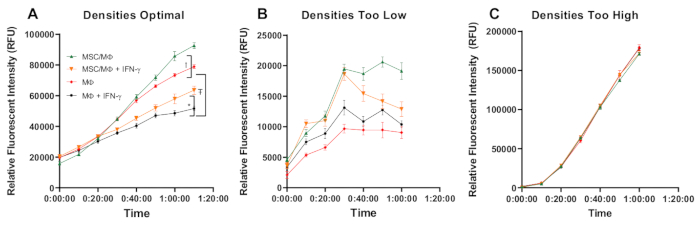
Figure 3: Representative quantitative data from the kinetic readdemonstrating optimal and suboptimal results. In (A), data are representative of an experiment with optimal MΦ cell density, in (B) data are representative of an experiment with suboptimal too low MΦ cell density, and in (C), data are representative of an experiment with suboptimal too high MΦ cell density. The relative fluorescence intensity measured in relative fluorescent units (RFU) is plotted on the Y-axis, while time is plotted on the X-axis. Note the differences in the range of RFU among optimal and sub-optimal experiments. In A, MSC partially rescue MΦ phagocytic activity in the setting of IFN-γ suppression over a 70 min period. The RFU is presented as mean ± the SEM, n = 6. The analysis was performed using Tukey's multiple comparisons test after a significant two-way ANOVA, Interaction effect P = 0.0001, Time effect P = 0.0001, and Treatment/MSC effect P = 0.0001. Symbols denote results of multiple comparison tests. * = significant difference between MSC/MΦ + IFN- γ vs MΦ + IFN-γ, Ŧ = significant difference between MΦ vs MΦ + IFN-γ, and † = significant difference between MSC/MΦ vs MΦ. See Table 3 for detailed results of the multiple comparison tests. Please click here to view a larger version of this figure.
Figure 4: Dynamic imaging videos providing visual confirmation of the cell-specific increase in fluorescence from acidic activation of labeled zymosan particles after incorporation into the MΦ phagolysosome. Time-lapse settings were acquisition every 1 min over a 30 min period using an exposure time of 400 ms and the EGFP filter set. (A) MΦ in monoculture, (B) MΦ in co-culture with MSC, (C) MΦ treated with IFN-γ (250 ng/mL) and (D) MΦ treated with IFN-γ (250 ng/mL) in co-culture with MSC. Please click here to download this File.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL | |||||
| B | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL | |||||
| C | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL | |||||
| D | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL | |||||
| E | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL | |||||
| F | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL | |||||
| G | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL | |||||
| H | CBL | MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ | CBL | RBL |
Table 1: Example of a 96-well plate design. CBL – Cell blank; MSC – seeded day 1; MΦ+ treated and MΦ untreated seeded day 2; RBL reagent blank added on assay day 3.
| 1 | 2 | 3 | 4 |
| MSC/MΦ | MSC/MΦ+ | MΦ | MΦ+ |
Table 2: Example of a 4-well chamber slide design. MSC – plated day 1; MΦ+ treated and MΦ untreated plated day 2.
| Tukey's multiple comparisons test | Mean Diff. | 95.00% CI of diff. | Below threshold? | Summary | Adjusted P Value |
| 0 minutes | |||||
| MSC/MΦ vs. MΦ | -3871 | -9495 to 1754 | No | ns | 0.2836 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 720.3 | -4904 to 6345 | No | ns | 0.9873 |
| MΦ vs. MΦ + IFN-γ | -77.17 | -5702 to 5548 | No | ns | >0.9999 |
| 10 minutes | |||||
| MSC/MΦ vs. MΦ | -3466 | -9091 to 2159 | No | ns | 0.3817 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 2326 | -3299 to 7950 | No | ns | 0.7062 |
| MΦ vs. MΦ + IFN-γ | 992 | -4633 to 6617 | No | ns | 0.968 |
| 20 minutes | |||||
| MSC/MΦ vs. MΦ | -1311 | -6936 to 4314 | No | ns | 0.9303 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 3315 | -2310 to 8940 | No | ns | 0.422 |
| MΦ vs. MΦ + IFN-γ | 3146 | -2478 to 8771 | No | ns | 0.4689 |
| 30 minutes | |||||
| MSC/MΦ vs. MΦ | 384.8 | -5240 to 6010 | No | ns | 0.998 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 2313 | -3312 to 7937 | No | ns | 0.7098 |
| MΦ vs. MΦ + IFN-γ | 8726 | 3101 to 14350 | Yes | *** | 0.0005 |
| 40 minutes | |||||
| MSC/MΦ vs. MΦ | 2247 | -3377 to 7872 | No | ns | 0.7278 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 4913 | -712.2 to 10537 | No | ns | 0.1101 |
| MΦ vs. MΦ + IFN-γ | 16521 | 10896 to 22145 | Yes | **** | <0.0001 |
| 50 minutes | |||||
| MSC/MΦ vs. MΦ | 5657 | 32.12 to 11282 | Yes | * | 0.0481 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 4932 | -692.9 to 10557 | No | ns | 0.1079 |
| MΦ vs. MΦ + IFN-γ | 19083 | 13458 to 24708 | Yes | **** | <0.0001 |
| 60 minutes | |||||
| MSC/MΦ vs. MΦ | 12376 | 6752 to 18001 | Yes | **** | <0.0001 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 9361 | 3736 to 14986 | Yes | *** | 0.0002 |
| MΦ vs. MΦ + IFN-γ | 24748 | 19123 to 30373 | Yes | **** | <0.0001 |
| 70 minutes | |||||
| MSC/MΦ vs. MΦ | 13770 | 8145 to 19395 | Yes | **** | <0.0001 |
| MSC/MΦ + IFN-γ vs. MΦ + IFN-γ | 11987 | 6362 to 17612 | Yes | **** | <0.0001 |
| MΦ vs. MΦ + IFN-γ | 27264 | 21639 to 32888 | Yes | **** | <0.0001 |
Table 3: Detailed statistical analysis of the data presented in Figure 3A. Results of Tukey's multiple comparison tests after a significant two-way ANOVA of data presented in Figure 3A.
Supplementary File 1: A representative spreadsheet file of raw kinetic data from an optimal experiment. Please click here to download this File.
