Modeling amyotrophic lateral sclerosis (ALS) in the laboratory is made uniquely challenging due to the overwhelmingly sporadic nature of over 80% of cases1, coupled with the vast number of genetic mutations known to be disease-causative2. Despite this, all cases of ALS share the unifying feature that before outright neuronal degeneration, there is dysfunctional communication between presynaptic motor neurons and postsynaptic muscle cells3,4. Clinically, as patients lose connectivity of the remaining upper and lower motor neurons, they present with features of neuronal hyper- and hypoexcitability throughout the disease5,6,7,8,9, reflecting complex underlying molecular changes to these synapses, which we, as ALS researchers, seek to understand.
Multiple transgenic models have illustrated that deterioration and disorganization of the neuromuscular junction occur with the expression of ALS-causative genetic mutations, including SOD110, FUS11,12, C9orf7213,14,15,16, and TDP4317,18,19 through morphological assessments, including evaluation of synaptic boutons, spine densities, and pre/postsynaptic organization. Mechanistically, since the landmark papers of Cole, Hodgkin, and Huxley in the 1930s, it has also been possible to evaluate synaptic responses through electrophysiological techniques in either in vitro cell culture or tissue slice preparations20. Through these strategies, many models of ALS have demonstrated synaptic transmission deficits. For example, a mutant variant of TDP43 causes enhanced firing frequency and decreases action potential threshold in NSC-34 (spinal cord x neuroblastoma hybrid cell line 34) motor-neuron-like cells21. This same variant also causes dysfunctional synaptic transmission at the neuromuscular junction (NMJ) before the onset of behavioral motor deficits in a mouse model22. It was previously showed that mutant FUS expression results in reduced synaptic transmission at the NMJ in a drosophila model of FUS-ALS before locomotor defects11. A recent report using induced pluripotent stem cells derived from C9orf72-expansion carriers revealed a reduction in the readily releasable pool of synaptic vesicles23. Altogether, these studies and others highlight the importance of building a more comprehensive understanding of the mechanisms underlying synaptic signaling in disease-relevant models of ALS. This will be pivotal in understanding the pathobiology of ALS and developing potential therapeutic targets for patients.
Methods of current and voltage clamping cells have been invaluable in determining membrane properties such as conductance, resting membrane potential, and quantal content of individual synapses20,24. However, one of the significant limitations of electrophysiology is that it is technically challenging and only provides insights from a single neuron at a time. Live-cell confocal microscopy, coupled with specific fluorescent probes, offers the opportunity to investigate the synaptic transmission of neurons in a spatiotemporal manner25,26,27. Although not a direct measure of neuronal excitability, this fluorescence approach can provide a relative measurement of two molecular correlations of synaptic function: synaptic vesicle release and calcium transients at synaptic terminals.
When an action potential reaches the presynaptic terminal region of neurons, calcium transients are triggered, facilitating the transition from an electrical signal to the process of neurotransmitter release28. Voltage-gated calcium channels localized to these areas tightly regulate calcium signaling to modulate the kinetics of neurotransmitter release29. The first reported fluorescence-based recordings of calcium transients were performed using either the dual-wavelength indicator Fura-2 AM or the single wavelength dye Fluo-3 AM30,31,32. While these dyes offered great new insight at the time, they suffer from several limitations such as non-specific compartmentalization within cells, active or passive dye loss from labeled cells, photobleaching, and toxicity if imaged over extended periods of time33. In the past decade, genetically encoded calcium indicators have become the workhorses for imaging various forms of neuronal activity. These indicators combine a modified fluorescent protein with a calcium chelator protein that rapidly switches fluorescence intensity after the binding of Ca2+ ions34. The application of these new indicators is vast, allowing for much easier visualization of intracellular calcium transients both in in vitro and in vivo settings. One family of these genetically encoded reporters, known as GCaMP, are now broadly utilized. These indicators contain a C-terminal calmodulin domain, followed by green fluorescent protein (GFP), and are capped by an N-terminal calmodulin-binding region35,36. Calcium-binding to the calmodulin domain triggers an interaction with the calmodulin-binding region, resulting in a conformational change in the overall protein structure and a substantial increase in the fluorescence of the GFP moiety35,36. Over the years, this family of reporters has undergone several evolutions to enable distinct readouts for particular calcium transients with specific kinetics (slow, medium, and fast), each with slightly different properties37,38. Here, the usage of the reporter GcaMP6 has been highlighted, which has been previously shown to detect single action potentials and dendritic calcium transients in neurons both in vivo and in vitro37.
Calcium transients in the presynaptic region trigger synaptic vesicle fusion events, causing neurotransmitter release into the synapse and initiation of signaling events in the postsynaptic cell28,39. Synaptic vesicles are both rapidly released and recycled, as the cell homeostatically maintains a stable cell membrane surface area and readily releasable pool of fusion capable membrane-bound vesicles40. The styryl dye used here has an affinity toward lipid membranes and specifically changes its emission properties based on the ordering of the surrounding lipid environment41,42. Thus, it is an ideal tool for labeling recycling synaptic vesicles and subsequent tracking of these vesicles as they are later released following neuronal stimulation41,42. The protocol that has been generated and optimized is an adaptation of the concepts described initially by Gaffield and colleagues, which allows us to visualize styryl dye-labeled synaptic vesicle puncta over time continuously41.
Here, two related fluorescence-based methodologies are described, reliably reporting specific cellular events involved in synaptic transmission. Protocols have been defined to probe the dynamics of depolarization-mediated presynaptic terminal calcium influx and synaptic vesicle exocytosis in cultured neurons. Here, methods and representative results are focused on using primary rodent cortical or motor neurons as the in vitro model system, as there are published studies using these cell types43,44. However, these methods are also applicable to differentiated human i3 cortical-like neurons45, as we have also had success with both protocols in presently ongoing experimentation in our laboratory. The general protocol is outlined in a stepwise linear format, shown in Figure 1. In brief, to study calcium dynamics in neurites, mature neurons are transfected with plasmid DNA to express the fluorescent reporter GCaMP6m under a Cytomegalovirus (CMV) promoter37,46. Transfected cells have a low level of basal green fluorescence, which increases in the presence of calcium. Regions of interest are specified to monitor fluorescence changes throughout our manipulation. This allows for highly spatially and temporally localized fluctuations in calcium to be measured37,46. For evaluating synaptic vesicle fusion and release, mature neurons are loaded with styryl dye incorporated into synaptic vesicle membranes as they are recycled, reformed, and reloaded with neurotransmitters in presynaptic cells41,42,43,47,48. The current dyes used for this purpose label synaptic vesicles along neurites and are used as a proxy for these regions in live-imaging experiments, as was shown by co-staining of styryl dye and synaptotagmin by Kraszewski and colleagues49. Included here are representative images of similar staining that have also been performed (Figure 2A). Previous investigators have extensively used such dyes to report synaptic vesicle dynamics at the neuromuscular junction and hippocampal neurons48,49,50,51,52,53,54,55,56. By selecting punctate regions of dye-loaded vesicles and by monitoring decreases in fluorescence intensity following vesicle release, functional synaptic transmission capacity and temporal dynamics of release can be studied following stimulation43. For both methods, a medium containing a high concentration of potassium chloride is employed to depolarize cells to mimic neuronal activity. Imaging parameters are specified to capture sub-second intervals spanning a baseline normalization followed by our stimulation capture period. Fluorescence measurements at each time point are determined, normalized to the background, and quantified over the experimental time period. Calcium-influx mediated GCaMP6m fluorescence increase or effective synaptic vesicle exocytosis styryl dye release fluorescence decrease can be detected through this strategy. Detailed methodological setup and parameters for these two protocols and a discussion on their advantages and limitations are described below.
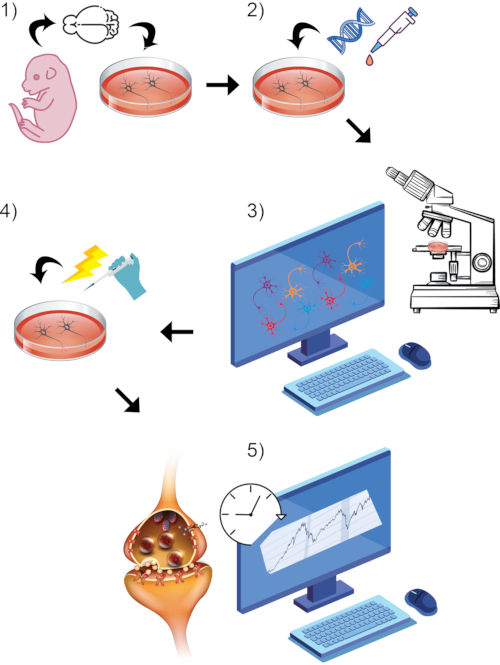
Figure 1: Visual rendering of overall general protocol process. (1) Isolate and culture primary rodent neurons in vitro to chosen maturation timepoint. (2) Introduce GCaMP DNA or styryl dye as reporters of synaptic activity. (3) Setup imaging paradigm using live-imaging equipped confocal microscope and associated software. Begin baseline recording period. (4) While cells are still undergoing live-image capture, stimulate neurons via high KCl bath perfusion. (5) Assess fluorescence intensity measurements over time to measure calcium transients or synaptic vesicle fusion. Please click here to view a larger version of this figure.
Following the successful implementation of the above protocol, representative results are shown for a typical styryl dye synaptic vesicle release experiment. Cultured rat primary cortical neurons were loaded with dye using the method described in section 6. The specificity of dye loading was determined by co-labeling with synaptic vesicle marker synaptophysin. A majority of styryl dye positive puncta are co-positive for this marker (Figure 2A). To determine whether the settings used for styryl dye imaging cause photobleaching, typically, an untreated well is loaded with dye and imaged for an extended 10 min period without stimulation to make sure fluorescence values remain constant.
Additionally, results are again shown using co-labeling of synaptophysin and styryl dye as in Figure 2A. These fluorophores were co-imaged using the paradigm indicated in section 6 for TRITC styryl dye imaging, plus dual capture of synaptophysin fluorescence using high-intensity laser power on the DAPI channel to visualize the mTurquoise-synaptophysin. Shown are representative images of synaptic regions pre- and post-stimulation (Figure 2B). While this method is an indirect inference for lack of photobleaching, analysis of the raw intensity values over the entire imaging period reveals that synaptophysin intensity remains constant, while styryl dye intensity goes down following stimulation (Figure 2C). Thus, taking this experiment and our other controls of stable fluorescence before stimulation and over extended periods in non-stimulation wells, we have established that decreases in styryl fluorescence can be attributed to synaptic unloading through KCl depolarization rather than photobleaching effects.
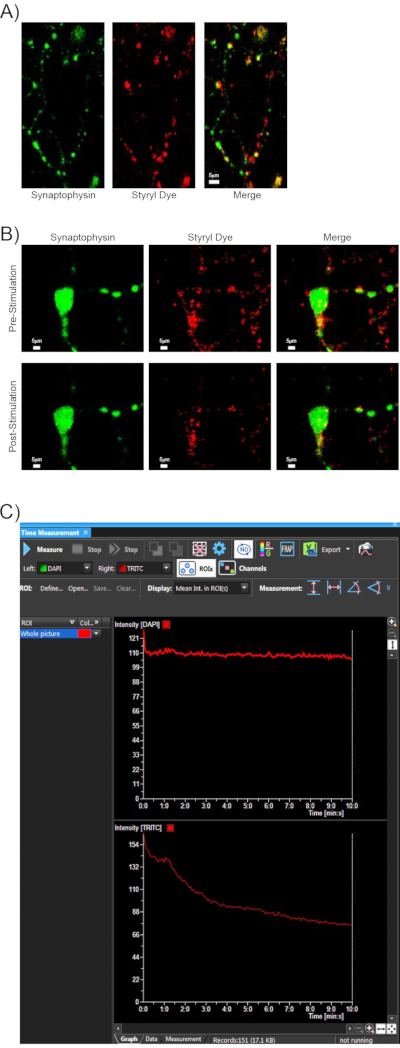
Figure 2: Assessment of specificity and imaging parameters of styryl dye labeling. (A) A representative 40x image of a styryl dye-labeled rat cortical neuron (red), which was transfected 24 h previously with a plasmid to express mTurquoise2-synaptophysin61 driven off the synapsin promoter (green). Colocalization of these two fluorophores indicates that a large majority of styryl dye positive puncta are co-stained with synaptic vesicle marker synaptophysin. Scale bar indicates 5 μm. (B) Cortical neurons were transfected and styryl dye loaded as in (A) and imaged according to the styryl dye paradigm along with dual fluorescence capture of the mTurquoise-synaptophysin on the DAPI channel. Representative images are showing synaptophysin and styryl dye puncta regions pre- and post-stimulation. Scale bar indicates 5 μm. (C) Fluorescence was monitored over time for synaptophysin (DAPI channel top) and styryl dye (FITC channel bottom). Synaptophysin intensity was maintained at a steady fluorescence over the imaging and stimulation period, while styryl dye fluorescence decreased as the dye was unloaded at stimulation. Please click here to view a larger version of this figure.
Representative images show the loading of styryl dye during imaging, with the selection of ROIs (Figure 3A–B). The raw fluorescence intensity of selected ROIs and a background ROI are plotted using the confocal software (Figure 3C). The neurons shown here were also transfected with plasmids to study the effects of dipeptide proteins produced in the context of C9ORF72-ALS, namely, FLAG-eGFP and FLAG-GA50-eGFP driven off of the T7 promoter. Successful synaptic vesicle release results in striking loss of dye fluorescence upon high KCl depolarization (Figure 3D, top two panels) for a GFP-transfected control neuron. A representative video included here demonstrates how this appears during imaging. In addition, a neuron selectively unloads styryl dye in a neurite region following stimulation (Video 1).
On the other hand, impaired synaptic transmission is represented by retained dye fluorescence even after high KCl depolarization in neurons transfected with a C9ORF72-linked dipeptide repeat construct (GA50) (Figure 3D, bottom two panels)43. Following quantitative data analysis (Figure 3E), data are represented as dye intensity throughout imaging for the control (GFP) and ALS/FTD (GA50) groups (Figure 3F)43. This method can also parse intermediate effects and is not simply an "all or nothing" binary measure. In experiments designed to rescue the synaptic deficits mediated by GA50, exogenous synaptic vesicle-associated protein 2 (SV2) was introduced by lentiviral transduction using the rSV2a-eGFP-pRRL plasmid driven off of the human PGK promoter. Following imaging and analysis as outlined above, synaptic firing was rescued in neurons co-expressing GA50 and SV2 (Figure 3G).
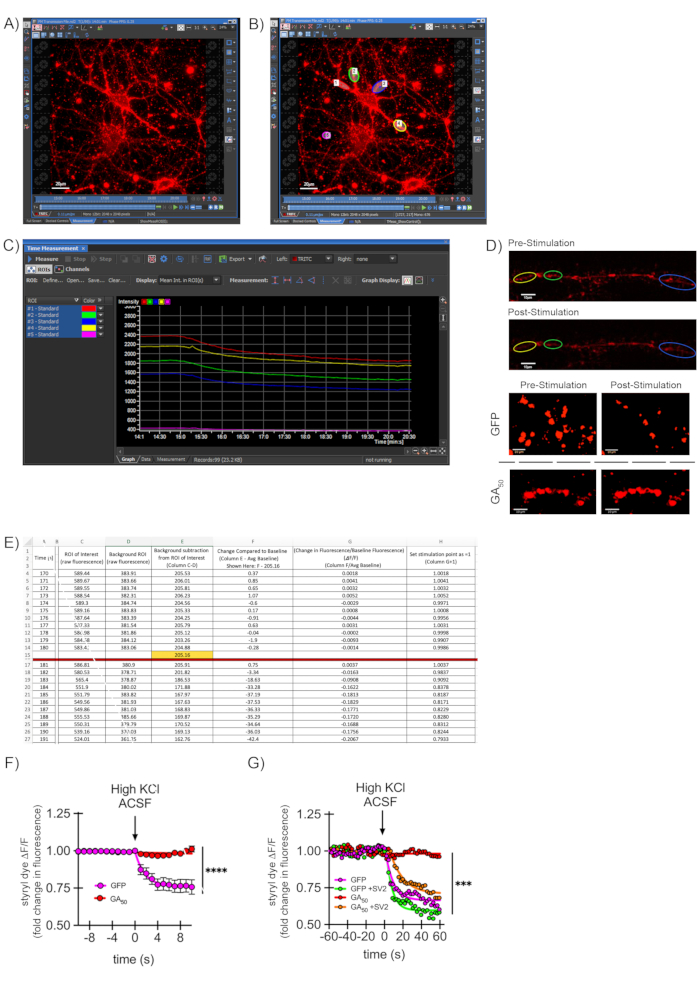
Figure 3: Evaluating synaptic vesicle release through styryl dye labeling. (A) A representative 40x image of a styryl dye-labeled neuron in low KCl aCSF media at the start of an imaging experiment. Scale bar indicates 20 μm. (B) Depiction of ROI generation on the image from (A). Specific puncta regions along neurites (#1-4) are chosen using the ROI tool, the bean-shaped button highlighted at the right. A final ROI (#5) is selected in a blank region to capture background signal intensity. Areas with non-specific, non-neuronal bright spots are avoided. Scale bar indicates 20 μm. (C) Representation of the signal intensities for ROIs #1-5 from (B) over time as graphed/depicted by confocal software. (D) Visual representations of ROI regions pre/post-stimulation for a neuron that effectively undergoes synaptic transmission (top two panels). The bottom two panels are zoomed images with an unbiased threshold applied to isolate puncta from the background to show distinct regions visually. GFP-containing neurons effectively undergo synaptic release, whereas expression of C9ORF2-ALS related peptide GA50 prevents this effect. Scale bars indicate 10 μm-Reprinted from Jensen et al. 202043. (E) Example of a quantification file using spreadsheet software, showing normalization of values to baseline and generation of ΔF/F values over time. Data are first normalized to the background ROI intensity value at each time point. This value is then compared to the average ROI of interest baseline value for the last 30 s pre-stimulation (shown here as 10 s in 1 s intervals for simplicity) (ΔF). This change is next calculated as a fraction of the baseline fluorescence (ΔF/F). Finally, the starting point value is set to 1 so that increases or decreases can be easily visualized graphically over time. At least five puncta regions per neuron and at least ten neurons per experimental condition are collected, with data such as this combined to generate average measurements at each time point. (F) Data from a spreadsheet file such as in (E) are moved into a graphing and statistics software program. For the graph shown here, the ΔF/F value at each timepoint averaged over all puncta regions of an experimental condition is plotted, along with its standard error of the mean. The graph here is data from the GFP versus GA50 experiments indicated in (D), where the last 10 s of baseline and first 10 s of the stimulation period are shown-reprinted from Jensen et al. 202043. (G) This assay can produce curves for intermediate synaptic release and is not simply an "all or nothing" response. GA50 was co-expressed with exogenous synaptic protein SV2 (synaptic vesicle-associated protein 2), and styryl dye imaging and analysis were performed as indicated in the previous steps. As in Figure 3F, the graph shown here represents the ΔF/F value at each timepoint averaged over all puncta regions of an experimental condition is plotted, along with its standard error of the mean. The graph shows the last minute of baseline and the first minute of the stimulation period from Jensen et al. 202043. Please click here to view a larger version of this figure.
Video 1: Representative video of styryl dye imaging experiment. Shown is a representative video of a cortical neuron loaded with styryl dye. The video shows the period beginning at the time of bath perfusion of high KCl aCSF. The boxed region highlights the neurites, with observable loss of puncta fluorescence over time. Please click here to download this Video.
Following the method described in section 7, shown are representative fluorescence images of cortical neurons transfected with GcaMP6m before and after KCl depolarization (Figure 4A). Raw intensity graphs show increased fluorescence values that fluctuate, indicating calcium entry into neurites following KCl-induced depolarization (Figure 4B). This second representative video shows neurons expressing GcaMP6m with low fluorescence at the end of the recording baseline period to demonstrate this effect. At the start of the stimulation, the neurons display a dramatic fluorescence increase (Video 2). This approach has been successfully utilized to demonstrate calcium-entry alterations in a mutant FUS-ALS model. Astrocytes in this model were transduced with adenovirus containing CMV-promoter-driven eGFP-FUS plasmids. When conditioned medium from mutant FUS-expressing astrocytes was placed on motor neurons, increased calcium influx was observed compared to non-mutant FUS conditions following α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) +cyclothiazide stimulation (Figure 4C)44. The sensitivity of this assay have been tested by performing experiments with and without calcium included in the perfusion medium. In the setting of GFP versus GA50 mentioned above, increased peak calcium transients was observed in GA50 containing motor neurons. Notably, this depended on calcium coming into the cell and not the release of internal calcium stores. When calcium was removed from the stimulating aCSF medium, no calcium transient responses were noted in either experimental condition (Figure 4D)43.
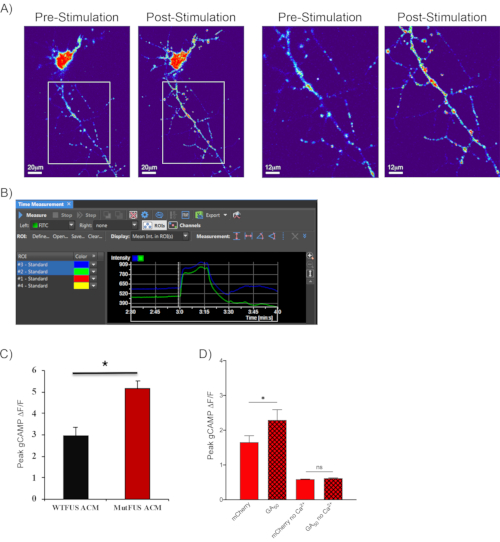
Figure 4: Observing calcium transients in real-time using GCaMP reporters. (A) A 40x image of a GCaMP6m expressing neuron, pseudo-colored to show the intensity of GFP fluorescence (low in blue to high in red). Left-hand panels show the whole 40x field of view pre- and post-stimulation. Scale bar indicates 20 μm. The images to the right are zoomed versions of the boxed areas revealed. Through these images, it is evident by eye that there is a robust increase in focal neuritic calcium levels following stimulation. Scale bar indicates 12 μm. (B) ROI selection and fluorescence intensity monitoring throughout the experiment is performed as in Figure 3. The final 30 s of baseline and 1 min stimulation for two selected ROIs of a neuron undergoing GCaMP6m fluorescence monitoring is shown here. In contrast to styryl dye imaging, the fluorescence intensity increases following neuronal stimulation. Additionally, as is noted here, calcium transients fluctuate in neurites over time; therefore, results are typically represented as the averaged peak normalized fluorescence change value for each ROI region. (C) Representative quantification of such an experiment is shown as was published in Kia-McAvoy et al. 201844, where primary motor neurons which received supernatant derived from mutant FUS-ALS astrocytes displayed significantly increased influx of calcium following AMPA receptor stimulation, compared to neurons receiving supernatant from wild-type FUS expressing astrocytes. (D) Representative quantification of calcium transient imaging where calcium was not included in the stimulating aCSF. Shown are conditions of mCherry versus GA50-mCherry stimulated with high KCl aCSF with or without calcium as published in Jensen et al. 202043. GA50 containing neurons displayed increased peak calcium influx compared with mCherry containing cells. There was no elevation of internal calcium levels in either experimental condition when calcium was removed from stimulating high KCL aCSF. Please click here to view a larger version of this figure.
Video 2: Representative video of GCaMP calcium transient experiment. Shown is a representative video of a field of cortical neurons transfected with GCaMP6m. Following a baseline period, a large calcium influx is noted at the 6 s mark when high KCl aCSF was applied. Calcium entry can be measured either for the whole cell, or at specific neurite regions as described. Please click here to download this Video.
| Potential problems | Possible reasons/solution | |||
| 1 | Non-specific loading | Be precise with the loading time. Longer loading times result in non-specific loading of FM4-64 into endosomes and lysosomes | ||
| 2 | No appreciable exocytosis in control neurons | Check the pH of high KCl aCSF solution. | ||
| 3 | Loss of focus and lateral drift | Make sure that perfect focus is on. Align the images post imaging using Nikon software or ImageJ plugin. | ||
| 4 | Bleaching of FM4-64 fluorescence | Check laser intensity used for imaging. Always try to use the lowest possible intensity. Confirm the laser intensity used does not result in bleaching by running trial runs. | ||
Table 2: Possible problems and troubleshooting for styryl dye experiments. Typical scenarios for issues and general troubleshooting for synaptic unloading experiments.
| Potential problems | Possible reasons/solution | |||
| 1 | Transfection efficiency too low | Check neuronal health. If neurons are hard to transfect one can switch to AAV or lentiviral transduction of GCaMP. | ||
| 2 | Accumulation of GCaMP6 fluorescence in the nucleus | Compromised neuronal health. Check transfection protocol. | ||
| 3 | Loss of focus and lateral Image drift | Make sure that perfect focus is on. Align the images post imaging using Nikon software or ImageJ plugin. | ||
| 4 | No calcium response upon KCL stimulation | Make sure the pH of aCSF is correct. | ||
| 5 | Bleaching of GCaMP6 fluorescence | Check laser intensity used for imaging. Always try to use the lowest possible intensity. Confirm the laser intensity used does not result in bleaching by running trial runs. | ||
| 6 | Blebbing of neurites | Compromised neuronal health due to transfection. Use a fresh batch of neuronal cultures. | ||
Table 3: Possible problems and troubleshooting for imaging calcium transients in neurites. Typical scenarios for issues and general troubleshooting for GCaMP calcium transient experiments.
