Single-Molecule Imaging of EWS-FLI1 Condensates Assembling on DNA
Summary
Here, we describe the use of the single-molecule imaging method, DNA Curtains, to study the biophysical mechanism of EWS-FLI1 condensates assembling on DNA.
Abstract
The fusion genes resulting from chromosomal translocation have been found in many solid tumors or leukemia. EWS-FLI1, which belongs to the FUS/EWS/TAF15 (FET) family of fusion oncoproteins, is one of the most frequently involved fusion genes in Ewing sarcoma. These FET family fusion proteins typically harbor a low-complexity domain (LCD) of FET protein at their N-terminus and a DNA-binding domain (DBD) at their C-terminus. EWS-FLI1 has been confirmed to form biomolecular condensates at its target binding loci due to LCD-LCD and LCD-DBD interactions, and these condensates can recruit RNA polymerase II to enhance gene transcription. However, how these condensates are assembled at their binding sites remains unclear. Recently, a single-molecule biophysics method-DNA Curtains-was applied to visualize these assembling processes of EWS-FLI1 condensates. Here, the detailed experimental protocol and data analysis approaches are discussed for the application of DNA Curtains in studying the biomolecular condensates assembling on target DNA.
Introduction
Transcriptional regulation is a crucial step for precise gene expression in living cells. Many factors, such as chromosomal modification, transcription factors (TFs), and non-coding RNAs, participate in this complicated process1,2,3. Among these factors, TFs contribute to the specificity of transcriptional regulation by recognizing and binding to specific DNA sequences known as promoters or enhancers and subsequently recruiting other functional proteins to activate or repress transcription4,5,6,7. How these TFs manage to search for their target sites in the human genome and interact with DNA coated with histones and non-histone DNA-binding proteins has perplexed scientists for decades. In the past few years, several classical models for the target search mechanism of TFs have been built to describe how they "slide," "hop," "jump," or "intersegment transfer" along the DNA chain8,9,10,11. These models are focused on the searching behavior on the DNA of one single TF molecule. However, recent studies show that some TFs undergo liquid-liquid phase separation (LLPS) either alone in the nucleus or with the Mediator complex12. The observed droplets of TFs are associated with the promoter or enhancer regions, highlighting the role of biomolecular condensate formation in transcription and the three-dimensional genome13,14,15. These biomolecular condensates are linked to membrane-lacking compartments in vivo and in vitro. They are formed via LLPS, in which modular biomacromolecules and intrinsically disordered regions (IDRs) of proteins are two main driving forces of multivalent interactions16. Thus, TFs not only search DNA but also function synergistically within these condensates4,17,18. To date, the biophysical property of these transcription condensates on DNA remains unclear.
Therefore, this study aimed to apply a single-molecule method-DNA Curtains-to directly image the formation and dynamics of the transcription condensates formed by TFs on DNA in vitro. DNA Curtains, a high-throughput in vitro imaging platform to study the interaction between proteins and DNA, has been applied in DNA repair19,20,21, target search22, and LLPS17,23,24. The flowcell of DNA Curtains is coated with biotinylated lipid bilayers to passivate the surface and allow the biomolecules to diffuse on the surface. The nanofabricated zig-zag patterns limit the movement of DNA. Biotinylated Lambda DNA substrates can align along the barrier edges and be stretched by the oriented buffer flow. The same starting and ending sequences of all the molecules allow the tracking of the protein on DNA and describe the position distribution of the binding events25,26. Moreover, the combination of DNA Curtains with total internal reflection fluorescence microscopy (TIRFM) helps minimize the background noise and detect signals at a single-molecule level. Thus, DNA Curtains could be a promising method to investigate the dynamics of transcription condensate formation on DNA motifs. This paper describes the example of an FUS/EWS/TAF15 (FET) family fusion oncoprotein, EWS-FLI1, generated by chromosomal translocation. Lambda DNA containing 25× GGAA-the binding sequence of EWS-FLI127– was used as the DNA substrate in the DNA Curtains experiments to observe how EWS-FLI1 molecules undergo LLPS on DNA. This manuscript discusses the experimental protocol and data analysis methods in detail.
Protocol
1. Preparation of the lipid bilayer master mix
- Rinse glass vials with double-distilled water (ddH2O) and 99% ethanol and dry them in a 60 °C drying oven.
- Make the lipid master mix by dissolving 1 g of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 100 mg of polyethylene glycol-reacted (PEGylated) lipids (18:1 of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (ammonium salt) (PEG2000 DOPE) and 25 mg of biotinylated lipids (18:1 of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt) (Biotinyl Cap DOPE)) into 10 mL of chloroform.
- Prepare 1 mL aliquots of the lipid master mix per vial and store them at -20 °C in clean glass vials.
NOTE: Store the dissolved lipid in glass vials and cover the vial cap with parafilm during storage.
2. Preparation of liposome solution
- Rinse a 2 mL glass vial with ddH2O and 99% ethanol and dry it thoroughly in a 60 °C drying oven.
- Rinse a 250 µL glass syringe with chloroform and then transfer 200 µL of the lipid master mix into the dry glass vial.
- Use a nitrogen spray gun to flow N2 gently while continuously rotating the vial in one direction.
NOTE: The N2 will help evaporate the chloroform, and the residual lipids will form a thin film on the wall of the glass vial. Adjust the nitrogen stream to be very gentle at first and increase the flow when a white film appears in the vial; complete this evaporation process within 5 min. - Transfer the glass vial to a vacuum drying oven at room temperature (RT) for 16-24 h for complete removal of the chloroform from the lipids.
- Add 2 mL of fresh lipid buffer (10 mM Tris-HCl (pH 7.5) and 100 mM NaCl) to the glass vial and dissolve the lipids at RT for 2 h. Vortex the solution several times and transfer it to a 5 mL polypropylene culture tube.
- Sonicate the lipid solution on ice with a micro-tip sonicator to form small unilamellar vesicles, using the following settings: 6 s on-12 s off (20% amplitude) for 6 cycles, then 6 s on-6 s off (30% amplitude) for 3 cycles, finally 10 s on (40% amplitude).
NOTE: The temperature increase during sonication could result in the bursting of foam and destroy the liposomes. Therefore, check the state of the foam and the temperature and replenish the ice frequently. - Filter the liposomes through a 0.22 µm nylon syringe filter, aliquot, and store it at 4 °C.
NOTE: As their mobility decreases with prolonged storage, the liposomes must be used in 2-3 months or prepared freshly before use.
3. Sequence cloning and biotinylation of Lambda DNA
- Insertion of the binding motifs into Lambda DNA
- Digest the target fragment containing the binding motifs and Lambda DNA with NheI and XhoI. Ligate the target fragment with Lambda DNA using T4 ligase overnight at RT.
- Culture E.coli VCS257 cells in antibiotic-free LB medium until the OD600 reaches 0.6. Store the bacteria at 4 °C for later use.
- Heat the ligation solution at 65 °C for 20 min to denature the T4 ligase.
- Thaw the Lambda phage extract, mix it with the ligated product gently by pipetting with blunt tips, and incubate the mixture at RT for 2 h.
- Add 500 µL of SM buffer (50 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 8 mM MgCl2, filtered through a 0.22 μm filter) and 20 µL of chloroform into the packaging mixture from step 3.1.4, mix well, and centrifuge the solution.
NOTE: The activity of the packaging extract can be maintained at 4 °C for prolonged periods. - Prepare the reaction mixture by mixing 10 µL of packaging extract with 90 µL of SM buffer and 100 µL of the VCS257 cells (from step 3.1.2) together, and incubate the mixture at 37 °C for 15 min.
NOTE: The packaging solution concentration depends on the density of the phage plaques on the following day; it can be diluted or added as needed. - Take ~5 mL of melted top agar (22 g/L NZCYM broth with 15 g/L agar) in a 15 mL tube. As soon as the top agar cools down to ~42 °C, add the reaction mixture (from step 3.1.6) slowly using a blunt-end pipette and pour the liquid onto an antibiotic-free LB agar plate. Keep the plate in a 37 °C incubator overnight.
- Confirm the positive plaques among the transparent phage plaques on the top agar plate by PCR. Transfer the positive plaques to a new top agar plate with a pipette and keep the plate in a 37 °C incubator overnight.
- Add 400 µL of VCS257 cells into 400 µL of buffer containing 10 mM MgCl2 and 10 mM CaCl2, inoculate one plaque into this cell mixture, and incubate it in a 37 °C shaker at 250 rpm for 15 min.
- Add 800 µL of this suspension containing the phage plaque to 200 mL of NZCYM broth in a 2 L flask and incubate it in a 37 °C shaker at 125 rpm.
- Measure OD600 of the bacterial suspension after ~4 h of cultivation. Keep in mind that the OD600 value will rise first and then drop to a trough at ~0.2. Stop the incubation when the OD600 value is about to rise again, add 500 μL of chloroform, and shake at 80 rpm for another 5 min.
NOTE: The increase in OD600 after the trough will occur quickly; hence, the OD600 value must be measured more often when it decreases to 0.3 to avoid excessive VCS257 cell growth in the culture. - Transfer the phage culture to a 500 mL bottle, add 11.7 g of NaCl, and shake the bottle. Incubate the bottle on ice for 10 min.
- Transfer the suspension equally into four 50 mL tubes. Centrifuge the tubes at 12,000 x g for 10 min.
NOTE: All these centrifugation steps in section 3.1 need to be done at 4 °C. - Transfer the supernatants to four new 50 mL tubes and centrifuge again at the same speed for 5 min.
- Collect the supernatants, add 10% (w/v) PEG8000 powder, and rotate at 4 °C to incubate for 30 min.
- Centrifuge the suspensions from step 3.1.15 at 12,000 x g for 15 min to obtain the phage pellets.
- Resuspend the pellets in 1 mL of phage dilution buffer (10 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 10 mM MgCl2) in four tubes and pour all 4 mL of these mixtures into a 50 mL tube.
- Add RNase A (10 mg/mL) and DNase I (4 mg/mL) to 20 µg/mL and 5 µg/mL final concentrations and incubate at 37 °C for 30 min to degrade excess nucleic acids.
- Add 4.5 mL of 300 mM Tris-HCl (pH 7.5), 2.76 mL of 0.5 M ethylenediamine tetraacetic acid (EDTA), 1.67 mL of 10% sodium dodecylsulfate (SDS), and 75 µL of proteinase K (10 mg/mL) into the tube, and incubate at 65 °C for 10 min.
- Add 4.5 mL of pre-cooled 3 M potassium acetate, and incubate on ice for 10 min.
- Centrifuge at 8,000 x g for 10 min, and spin the supernatant for an additional 5 min at 10,000 x g.
- Add 70% volume of isopropanol into the collected supernatant (from step 3.1.21) with sufficient mixing, incubate at RT for 2 min, and then centrifuge at 8,000 x g for 10 min. Discard the isopropanol and spin again for 5 min.
NOTE: The DNA pellet will be smeared before the first 10 min centrifugation; the pellet will be concentrated at the bottom of the tube after the second 5 min centrifugation. - Wash the precipitated DNA with 2-3 mL of 70% ethanol and spin down again for 1 min. Dry the pellet at 4 °C for 2 h, and then resuspend the DNA in 500 µL of TE buffer (10 mM Tris-HCl (pH 8.0) and 1 mM EDTA) overnight at RT.
- Transfer the DNA to a 1.5 mL tube and spin it at 18,000 x g for 3 min. Transfer the DNA to a new 1.5 mL tube. Aliquot the purified Lambda DNA and store it at -20 °C.
- Biotin-Lambda DNA preparation
- Melt 50 µL of purified Lambda DNA with cloned motifs at 65 °C for 10 min.
NOTE: As Lambda DNA is ~48 kbp long, it must be heated before pipetting to avoid DNA breaks. - Mix 50 µL of Lambda DNA, 1 µL of Biotin primer (5' – (Phos) – AGG TCG CCG CCC – BIOTEG – 3') (100 µM) and 54 µL of ddH2O together. Co-incubate the mixture at 65 °C for 5 min and leave it on the bench to cool down to RT.
- Add 12 µL of 10x T4 ligase buffer and 3 µL of T4 ligase, and incubate at RT for several hours or overnight.
- Add an equal volume of phenol-chloroform (1:1) mixture to the ligation product, mix well immediately, and centrifuge at 12,000 x g for 2 min at RT.
- Transfer the upper aqueous phase containing the DNA carefully to a new tube, add an equal volume of chloroform, mix well, and then centrifuge at 12,000 x g for 2 min.
- Transfer the upper aqueous phase to a new tube, add an equal volume of isopropanol and 10% of the volume of 3 M sodium acetate, and incubate at RT for 2 min. Centrifuge the mixture at 12,000 x g for 5 min.
- Remove the supernatant, wash the pellet with 75% ethanol, and then centrifuge at 12,000 x g for 5 min.
- Remove the 75% ethanol, air-dry the DNA at RT for 10 min, and use 120 µL of TE buffer to dissolve the DNA.
- Add 60 µL of Buffer A (30% (w/v) PEG8000 and 30 mM MgCl2) to 120 µL of the solution from step 3.2.8, and rotate at 4 °C for 20-24 h.
- Centrifuge the mixture at 18,000 g for 5 min, and remove the supernatant.
- Use 180 µL of pre-cooled 75% ethanol to wash the pellet, and repeat step 3.2.10.
- Dry the pellet at RT, and use 100 µL of TE150 buffer to dissolve the DNA.
- Melt 50 µL of purified Lambda DNA with cloned motifs at 65 °C for 10 min.
4. Nanofabricated zig-zag barriers
- Clean slides in acetone for 20 min by sonication and transfer them to a new glass container with 1 M NaOH for sonication for another 20 min. Mix 90 mL of H2SO4 with 30 mL of H2O2, and immerse the slides in this mixture for 30 min. Rinse the slides with acetone and isopropanol and dry the slides with N2.
NOTE: Mixing H2SO4 and H2O2 is an exothermic process; take adequate safety precautions during the operation. - Coat the cleaned slide with polymethylmethacrylate (PMMA) 25 kDa, PMMA 49.5 kDa, and the top layer of conductive protective coating in succession. Use electron beam lithography to write the zig-zag barriers (Figure 1A).
- Wash the slides with ddH2O to remove the conductive protective coating and dry the slides with N2.
- Deposit a 30 nm layer of chromium (Cr) using a magnetron sputtering machine and remove the remaining PMMA layers with acetone.
NOTE: One flowcell can be reused for more than 20 DNA Curtains experiments. To reuse the flowcell, wash it with ethanol, detergent, and NaOH.
5. Purification of EWS-FLI1 protein
NOTE: The observation of 500 nM EWS-FLI1 on Lambda DNA with 25× GGAA motifs serves as a good example for the application of DNA Curtains to condensate formation. EWS-FLI1 is a fusion protein combining the N-terminal of EWSR1 (1-265) and the C-terminal of FLI1 (220-453). An mCherry tag was fused to the N-terminal of the EWS-FLI1 fusion protein for visualization.
- Clone the EWS-FLI1 gene into a reconstituted pRSF vector or any other suitable prokaryote expression vector.
- Transform the plasmid encoding 7x His-mCherry-EWSFLI1 into the E.coli BL21(DE3) strain; grow a single colony in 5 mL of LB medium at 37 °C overnight. When the OD600 value reaches 0.6-0.8 after transferring to 2 L of LB medium, add 0.5 mM IPTG and shake the cell culture at 18 °C for 16-18 h.
- Centrifuge the culture to remove the supernatant and resuspend the cells with lysis buffer (50 mM Tris-HCl, pH 7.5; 1 M KCl; 1 M urea; 10 mM imidazole; 1.5 mM beta-mercaptoethanol (β-ME); and 5% glycerol).
- After sonication and centrifugation at 18000 × g for ~30 min, load the supernatant onto Ni-NTA resin equilibrated in a 5-fold volume of lysis buffer, and then wash the resin with a 10-fold volume of washing buffer (50 mM Tris-HCl (pH 7.5), 1 M KCl, 1 M urea, 40 mM imidazole, 1.5 mM β-ME, and 5% glycerol).
- Elute the bound protein with 15 mL of elution buffer (50 mM Tris-HCl (pH 7.5), 1 M KCl, 1 M urea, 500 mM imidazole, 1.5 mM β-ME, and 5% glycerol) and concentrate the elution to ~500 µL.
- Load the concentrated protein solution onto a gel filtration column and analyze the products by SDS-polyacrylamide gel electrophoresis. Quickly freeze the purified protein in liquid nitrogen and store at -80 °C.
6. Preparation of a DNA Curtains flowcell
- Prepare a flowcell with zig-zag nanofabricated barriers for a DNA Curtains experiment and determine the direction of flow. To do this, connect the input and output tubes in the correct direction.
- Wash the flowcell with 2.5 mL of lipid buffer using two 3 mL syringes. Ensure there is no bubble in the flowcell.
- Remove the syringe from the output, and inject 1 mL of the liposome solution (40 µL of liposome stock from section 2 and 960 µL of lipid buffer) three times with 5 min incubation after each injection.
- For the healing step, wash the flowcell with 2.5 mL of lipid buffer slowly, and incubate at RT for 30 min.
NOTE: The healing time can be longer if necessary; if any bubbles appear, perform double-healing by repeating steps 5.3 and 5.4 to remove the bubbles. - Prepare 30 mL of bovine serum albumin (BSA) buffer (40 mM Tris-HCl (pH 7.5), 2 mM MgCl2, 1 mM dithiothreitol, and 0.5 mg/mL BSA for surface passivation). After 30 min of healing, wash the flowcell with 2.5 mL of BSA buffer from the output side.
NOTE: The BSA stock is a 20 mg/mL solution stored at 4 °C and must be used within 1-2 weeks. The BSA concentration in the buffer can be adjusted according to the surface condition. - Inject 800 µL of streptavidin buffer (10 µL of a 1 mg/mL streptavidin stock, 790 µL of BSA buffer) into the flowcell from the input side in two steps with 10 min of incubation after each step.
NOTE: The streptavidin anchors the DNA onto the lipids due to its multivalency by bridging between the biotinylated lipids and the biotinylated DNA. - Wash the flowcell with 2.5 mL of BSA buffer to remove all free streptavidin molecules.
- Dilute the Biotin-Lambda DNA with cloned motifs from section 3 using BSA buffer (2.5 µL of DNA and 998 µL of BSA buffer). Inject Lambda DNA 4 times slowly at 5 min intervals.
- Turn on the microscope and the scientific Complementary Metal Oxide Semiconductor (sCMOS) system during the injection. Wash the tubing with 10 mL of ddH2O. Rinse the prism and the tubing connector with water, 2% liquid cuvette cleaner, and 99% ethanol. Prepare the imaging buffer by adding KCl and double-stranded DNA dye into the BSA buffer to obtain final concentrations of 150 mM and 0.5 nM, respectively, and take up at least 20 mL of the imaging buffer into a 30 mL syringe.
- Set up the flowcell on the microscope stage and connect it to the microfluidic system.
- Use a flow rate of 0.03 mL/min to flush the DNA molecules to the barrier for 10 min, incubate for 30 min with the flow stopped, and then switch it off to let the DNA diffuse laterally.
NOTE: The purpose of this 30 min incubation is to let the DNA molecule distribute more evenly in front of the barrier through simple diffusion because DNA molecules tend to accumulate on the side of the barrier that is closed to the input where they are flushed in. The incubation time can be adjusted as necessary.
7. Imaging of EWS-FLI1 condensation formation on DNA Curtains
- Open the imaging software, find and mark the positions of the 3 × 3 zig-zag patterns under bright-field.
- Turn on the flow at 0.2 mL/min to stain the DNA with the double-stranded DNA dye for 10 min.
- Dilute mCherry-EWS-FLI1 with the imaging buffer to the concentration of 100 nM in 100 µL in the tube.
- Load the protein sample through the valve with a 100 µL glass syringe, and change the flow rate to 0.4 mL/min.
- Turn on the 488 nm laser and pre-scan each region to check the DNA distribution state; select the region in which the DNA molecules distribute evenly. Set the laser power to 10% for the 488 nm laser and 20% for the 561 nm laser. Use the power meter to measure the real laser power near the prism: 4.5 mW for the 488 nm laser and 16.0 mW for the 561 nm laser.
- Image acquisition
- Start acquiring images at 2 s intervals with both 488 nm and 561 nm lasers simultaneously.
- Change the valve from manual mode to injection mode to let the imaging buffer flush the protein sample into the flowcell after 60 s.
NOTE: This process will take ~30 s, and the field-of-view will be covered with mCherry signals as soon as the EWS-FLI1 proteins reach the flowcell. - To remove free EWS-FLI1, keep washing the flowcell with the imaging buffer for 5 min with only the 561 nm laser switched on. Stop the flow and incubate at 37 °C for 10 min.
- Turn on the flow at 0.4 mL/min to let the DNA extend, and acquire images at 2 s intervals between different frames to obtain high-throughput data of EWS-FLI1 condensate formation.
8. Intensity analysis for mCherry-EWS-FLI1
- Import the data as image sequences into ImageJ software, pick out the puncta at 25× GGAA sites in an 8× 8 pixels square, and save the image in .tif format.
- Calculate the intensity as the summation of the intensity in the 8×8 pixels square, including the whole puncta, and remove the background by subtracting the intensity of background in an area of the same size near the 8 x 8 pixels square.
Representative Results
The schematic of DNA Curtains is shown in Figure 1A, Figure 1B, and Figure 1D. The cloned target sequence containing 25 uninterrupted repeats of GGAA is found in the NORB1 promoter in Ewing sarcoma. This target sequence is crucial for EWS-FLI1 recruitment28. EWS-FLI1 molecules were visualized by detecting the mCherry-labeled EWS-FLI1 signals obtained with a 561 nm laser (Figure 1C and Figure 1E). After a DNA Curtains experiment was set up, the in vitro formation of biomolecular condensates of EWS-FLI1 at the 25× GGAA sites of the DNA substrate could be directly visualized (Figure 1B–E). The specificity of the mCherry-EWS-FLI1 used in DNA Curtains was confirmed by an electrophoretic mobility shift assay using a DNA template containing 25× GGAA and without GGAA separately (Figure 2A). Additionally, the concentration of EWS-FLI1 was titrated from 20 nM to 500 nM, and the intensity of the puncta at the 25× GGAA sites was determined. Compared with the intensity of mCherry-FLI1DBD, the intensity of EWS-FLI1 increased dramatically, whereas the change in the intensity of FLI1DBD was negligible when the proteins were saturated to cover the 25× GGAA sites. Therefore, these results strongly suggest that EWS-FLI1 formed condensates on DNA (Figure 2B–E).
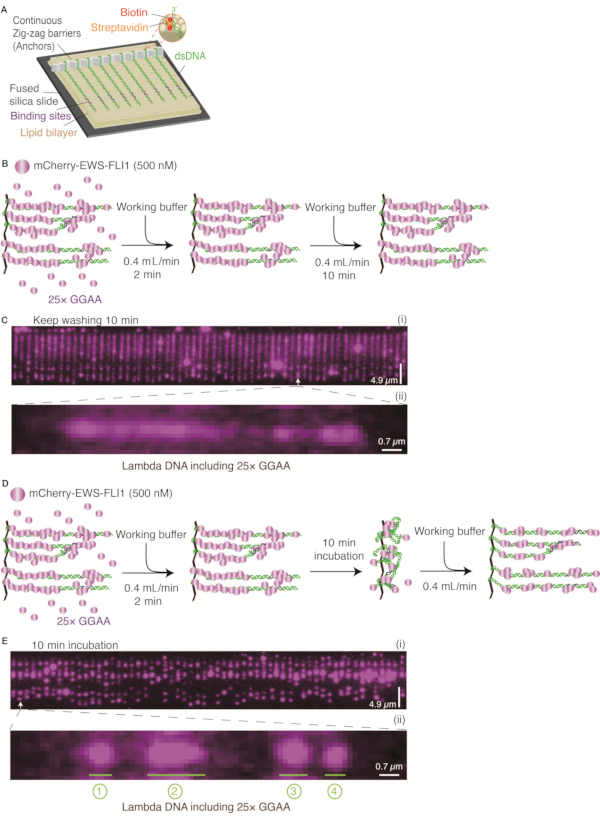
Figure 1: EWS-FLI1 condensate formation on Lambda DNA containing 25× GGAA motifs. (A) Schematic of DNA Curtains. (B and D) Two strategies for detecting EWS-FLI1 condensates (500 nM) assembling on Lambda DNA: (B) Keep washing for 10 min;(D)incubate for 10 min. (C and E) Representative wide-field total internal reflection fluorescence microscopy image of DNA Curtains: (C) Detection strategy in B; (E) Detection strategy in D. C (ii) and E (ii) DNA is zoomed in to show the distinct puncta formed on a single DNA substrate. DNA substrates are Lambda DNA with 25× GGAA motifs. Numbers "1, 2, 3, 4" represent different positions of puncta on one Lambda DNA, and "3" is where the 25× GGAA microsatellite sequence was cloned. Scale bars = 4.9 µm (C, E (i)) and 0.7 µm (C, E (ii)). This figure has been modified from 24. Abbreviation: dsDNA = double-stranded DNA. Please click here to view a larger version of this figure.
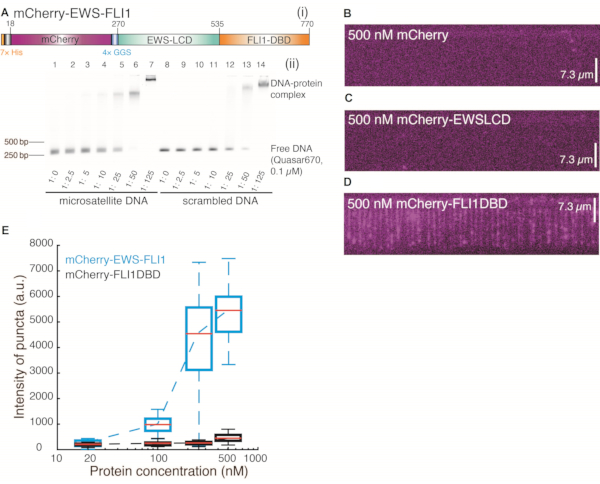
Figure 2: Binding events of the detached domain of mCherry-EWS-FLI1 on 25× GGAA repeats. (A) (i) mCherry-EWS-FLI1. (ii) Electrophoretic mobility shift assay of mCherry-EWS-FLI1: 306-bp dsDNA labeled with 5' Quasar670 was incubated with mCherry-EWS-FLI1 at different concentrations under room temperature for 30 min in reaction buffer containing 40 mM Tris-HCl (pH 7.5), 150 mM KCl, 2 mM MgCl2, 1 mM DTT, and 0.2 mg/mL bovine serum albumin. The samples were loaded and run on 1.3% agarose gel for 25 min, 120 V. (B–D) Wide-field total internal reflection fluorescence microscopy images of 25× GGAA with DNA Curtains after incubation with 500 nM mCherry (B), 500 nM mCherry-EWSLCD (C), or 500 nM mCherry-FLI1DBD (D). (E) Intensity distribution of EWSFLI1 (cyan) and FLI1DBD (black) signals at the target site (25× GGAA) region vs. protein concentration. This figure has been modified from 24. Abbreviations: LCD = low-complexity domain; DBD = DNA-binding domain. Please click here to view a larger version of this figure.
Discussion
As single-molecule approaches are extremely sensitive to the contents of the reaction system, extra effort must be invested to ensure good quality of all the materials and solutions during the DNA Curtains experiments, especially the lipids prepared in sections 1 and 2 and the buffers used in section 5. Reagents of higher purity must be used to prepare buffers, and buffers must be freshly prepared for the single-molecule assay
When 500 nM mCherry-labeled EWS-FLI1 was flushed into the chamber, several magenta puncta appeared on Lambda DNA containing the 25× GGAA sequences. Notice that there was a consecutive non-specific distribution of magenta signals throughout the entire DNA even after 10 min of washing with blank buffer (Figure 1B,C). Interestingly, EWS-FLI1 molecules were rearranged on DNA during the 10 min incubation without the buffer flow and gathered into several puncta (Figure 1D,E). One of these puncta was formed at the cloned 25× GGAA site, while all others were formed in the regions containing a high density of consecutive GGAA motifs. This phenomenon strongly suggests that the no-flow incubation procedure allows EWS-FLI1 molecules to search the loci and assemble on DNA faster.
Several control experiments must be performed to clarify how these condensates formed on DNA Curtains. We purified mCherry, mCherry-EWSLCD, and mCherry-FLI1DBD and followed the same procedure to inject these proteins into the chamber. Neither mCherry (Figure 2B) nor mCherry-EWSLCD (Figure 2C) left any signals on DNA, indicating that the FLI1DBD of EWS-FLI1 was necessary for the interaction with DNA. To confirm that phase separation occurred in DNA Curtains at such low protein concentrations, the concentration of mCherry-EWS-FLI1 was titrated from 25 nM to 500 nM, and the intensity of each puncta on one DNA molecule was determined at the clone site (Figure 2E). A comparison with the intensity of fusion TF FLI1DBD labeled with mCherry revealed that although the puncta intensities of EWS-FLI1 and FLI1-DBD were similar at low protein concentrations, the intensity of EWS-FLI1 increased dramatically while that of FLI1DBD remained low even when the concentration reached 500 nM. These results suggest that EWS-FLI1 molecules bind to the 25× GGAA sequence and assemble into a condensate on it through LCD interactions. A single FLI1DBD can bind 2x GGAA motifs, and higher-order oligomers bind to highly repetitive low-affinity sequences27. The mCherry-FLI1DBD signal on the 25× GGAA sequence was from a protein cluster rather than an individual protein molecule. Although mCherry-FLI1DBD could bind the 25× GGAA sites, it failed to assemble as a condensate on DNA, confirming that the LCD-LCD interaction was necessary for phase separation (Figure 2D,E).
Single-molecule methods enable researchers to study the dynamics inside transcription factor condensates. The DNA Curtains method has some advantages compared with other single-molecule methods such as single-molecule fluorescence resonance energy transfer (smFRET)29, super-resolution imaging30, and optical tweezer31,32. First, the DNA Curtains method allows for the reconstitution of the transcription machinery on long genomic DNA in vitro and the real-time observation of transcription condensate formation with high-throughput data acquisition. Second, aligned DNA molecules allow the mapping of the position of condensates on each DNA strand. Thus, the preferred DNA sequences for puncta formation can be easily determined.
Moreover, long-term acquisition is feasible with DNA Curtains, allowing for the measurement of the on-rate (kon) and off-rate (koff) of one punctum. Nevertheless, DNA Curtains has some inherent technical defects, necessitating the evidence from different methods to be collectively examined. On the one hand, the resolution of DNA Curtains can only reach 0.18 µm or ~1,000-bp because the long Lambda DNA template has a restriction with respect to the diffraction limit, which can hinder the differentiation of two neighboring fluorescence signals. On the other hand, the flow is used to extend double-strand DNA (dsDNA), and the force applied on the biomolecule may influence the diffusion property of the proteins binding to the DNA. Double-tethered DNA Curtains can anchor both ends of dsDNA and record the movement of proteins without flow, which is a noteworthy solution33. To summarize, deepening our understanding of the dynamic assembly of biomolecular condensates on DNA in real-time will shed light on not only the biophysical mechanism of LLPS but also on the basic biology of LLPS-related cellular processes, such as gene transcription regulation24.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by NSFC Grants No. 31670762 (Z.Q.).
Materials
| 488 nm diodepumped solid-state laser | Coherent | OBIS488LS | |
| 561 nm diodepumped solid-state laser | Coherent | OBIS561LS | |
| Agar | Rhawn | R003215-50g | |
| biotinylated DOPE | Avanti | 870273P | |
| Bovine Serum Albumin | Sigma | A7030 | |
| Chloroform | Amresco | 1595C027 | |
| Coating Electra 92 | Allresist GmbH | AR-PC 5090.02 | The conductive protective coating |
| Deoxyribonuclease I bovine | Sigma | D5139-2MG | |
| DOPC | Avanti | 850375P | |
| DTT | Sigma | D9779 | |
| Glass coverslip | Fisher Scientific | 12-544-7 | |
| Hellmanex III | Sigma | Z805939-1EA | |
| KCl | Sigma | 60130 | |
| Lambda DNA | NEB | N3013S | |
| Lambda Packing Extracts | Epicentre | MP5120 | |
| MgCl2 | Sigma | M2670 | |
| NaCl | Sigma | s3014 | |
| Nanoport | Idex | N-333-01 | |
| NheI-HF | NEB | R3131S | |
| Nikon Inverted Microscope | Nikon | Eclipse Ti | |
| NZCYM Broth | Sigma | N3643-250G | |
| PEG-2000 DOPE | Avanti | 880130P-1G | |
| PEG-8000 | Amresco | 25322-68-3 | |
| PMMA 200K, ETHYL LACTATE 4% | Allresist GmbH | AR-P 649.04 | |
| PMMA 950K, ANISOLE 2% | Allresist GmbH | AR-P 672.02 | |
| Prime 95B Scientific CMOS camera | PHOTOMETRICS | Prime95B | |
| proteinase K | NEB | P8107S | |
| Silica glass slide | G.Finkenbeiner | ||
| Six-way injection valve | Idex | MXP9900-000 | |
| Streptavidin | Thermo | S888 | Diluted with ddH2O |
| Syringe pump | Harvard Apparatus | Pump11 Elite | |
| T4 DNA Ligase | NEB | M0202S | |
| Tris base | Sigma | T6066 | |
| XhoI | NEB | R0146V | |
| YOYO-1 Iodide (491/509) | Invitrogen | Y3601 | Diluted with DMSO |
Referências
- Cramer, P. Organization and regulation of gene transcription. Nature. 573 (7772), 45-54 (2019).
- Zhang, Y., Reinberg, D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes & Development. 15 (18), 2343-2360 (2001).
- Wang, X., et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 454 (7200), 126-130 (2008).
- Alexander, K. A., et al. p53 mediates target gene association with nuclear speckles for amplified RNA expression. Molecular Cell. 81 (8), 1666-1681 (2021).
- Matsui, T., Segall, J., Weil, P. A., Roeder, R. G. Multiple factors required for accurate initiation of transcription by purified RNA polymerase-II. Journal of Biological Chemistry. 255 (24), 1992-1996 (1980).
- Stadhouders, R., Filion, G. J., Graf, T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature. 569 (7756), 345-354 (2019).
- Peng, Y., Zhang, Y. Enhancer and super-enhancer: Positive regulators in gene transcription. Animal Models and Experimental Medicine. 1 (3), 169-179 (2018).
- Lambert, S. A., et al. The human transcription factors. Cell. 172 (4), 650-665 (2018).
- Normanno, D., Dahan, M., Darzacq, X. Intra-nuclear mobility and target search mechanisms of transcription factors: a single-molecule perspective on gene expression. Biochimica et Biophysica Acta. 1819 (6), 482-493 (2012).
- Berg, O. G., Winter, R. B., von Hippel, P. H. Diffusion-driven mechanisms of protein translocation on nucleic acids. Bioquímica. 20 (24), 6929-6948 (1981).
- Loverdo, C., et al. Quantifying hopping and jumping in facilitated diffusion of DNA-binding proteins. Physical Review Letters. 102 (18), 188101 (2009).
- Boija, A., et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 175 (7), 1842-1855 (2018).
- Sabari, B. R., et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 361 (6400), (2018).
- Kim, S., Shendure, J. Mechanisms of interplay between transcription factors and the 3D genome. Molecular Cell. 76 (2), 306-319 (2019).
- Shrinivas, K., et al. Enhancer features that drive formation of transcriptional condensates. Molecular Cell. 75 (3), 549-561 (2019).
- Banani, S. F., Lee, H. O., Hyman, A. A., Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nature Reviews. Molecular Cell Biology. 18 (5), 285-298 (2017).
- Zhou, H., et al. Mechanism of DNA-induced phase separation for transcriptional repressor VRN1. Angewandte Chemie International Edition. 58 (15), 4858-4862 (2019).
- Rao, S., Ahmad, K., Ramachandran, S. Cooperative binding between distant transcription factors is a hallmark of active enhancers. Molecular Cell. 81 (8), 1651-1665 (2021).
- Qi, Z., et al. DNA sequence alignment by microhomology sampling during homologous recombination. Cell. 160 (5), 856-869 (2015).
- Qi, Z., Greene, E. C. Visualizing recombination intermediates with single-stranded DNA curtains. Methods. 105, 62-74 (2016).
- Ma, C. J., Gibb, B., Kwon, Y., Sung, P., Greene, E. C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Research. 45 (2), 749-761 (2017).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507 (7490), 62-67 (2014).
- Larson, A. G., et al. Liquid droplet formation by HP1 alpha suggests a role for phase separation in heterochromatin. Nature. 547, 236-240 (2017).
- Zuo, L., et al. Loci-specific phase separation of FET fusion oncoproteins promotes gene transcription. Nature Communications. 12 (1), 1491 (2021).
- Collins, B. E., Ye, L. F., Duzdevich, D., Greene, E. C. DNA curtains: novel tools for imaging protein-nucleic acid interactions at the single-molecule level. Methods in Cell Biology. 123, 217-234 (2014).
- Greene, E. C., Wind, S., Fazio, T., Gorman, J., Visnapuu, M. -. L. DNA curtains for high-throughput single-molecule optical imaging. Methods in Enzymology. 472, 293-315 (2010).
- Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., Lessnick, S. L. Emergent properties of EWS/FLI regulation via GGAA microsatellites in Ewing’s sarcoma. Genes & Cancer. 1 (2), 177-187 (2010).
- Sizemore, G. M., Pitarresi, J. R., Balakrishnan, S., Ostrowski, M. C. The ETS family of oncogenic transcription factors in solid tumours. Nature Reviews. Cancer. 17 (6), 337-351 (2017).
- Roy, R., Hohng, S., Ha, T. A practical guide to single-molecule FRET. Nature Methods. 5 (6), 507-516 (2008).
- Huang, B., Bates, M., Zhuang, X. W. Super-resolution fluorescence microscopy. Annual Review of Biochemistry. 78, 993-1016 (2009).
- Bustamante, C., Bryant, Z., Smith, S. B. Ten years of tension: single-molecule DNA mechanics. Nature. 421 (6921), 423-427 (2003).
- Bustamante, C., Chemla, Y. R., Moffitt, J. R., Selvin, P. R., Ha, T. High-resolution dual-trap optical tweezers with differential detection. Single-Molecule Techniques: A Laboratory Manual. , (2008).
- Zhao, Y. L., Jiang, Y. Z., Qi, Z. Visualizing biological reaction intermediates with DNA curtains. Journal of Physics D: Applied Physics. 50 (15), 153001 (2017).
