The caspases are a family of cysteine aspartate proteases that can be grouped into initiator caspases and executioner caspases. Executioner caspases comprise caspase-3, -6 and -7. They are naturally found in cells as dimers and are cleaved by the initiator caspases to execute apoptosis1. Initiator caspases include human caspase-1, -2, -4, -5, -8, -9, -10 and -12. They are found as inactive zymogens (pro-caspases) that are activated by proximity-induced dimerization and stabilized by auto-proteolytic cleavage2,3. The inflammatory caspases are a subset of the initiator caspases2 and encompass caspase-1, -4, -5, and -12 in humans, and caspase-1, -11, and -12 in mouse4,5. Rather than an apoptotic role, they play a central role in inflammation. They mediate proteolytic processing and secretion of pro-interleukin (IL)-1β and pro-IL-186,7, which are the first cytokines to be released in response to pathogenic invaders8,9. Caspase-1 is activated upon recruitment to its activation platform; a large molecular weight protein complex termed the inflammasome (Figure 1A)10. Dimerization of caspase-4, -5, and -11 occurs independently of these platforms through a noncanonical inflammasome pathway11,12.
Canonical inflammasomes are cytosolic multimeric protein complexes that consist of an inflammasome sensor protein, the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), and the effector protein caspase-110. The most well studied canonical inflammasomes are the NOD-like receptor family containing a pyrin domain (NLRP), NLRP1 and NLRP3, the NLR family containing a CARD (NLRC), NLRC4, and the absent in melanoma 2 (AIM2). They each contain a pyrin domain, a CARD, or both domains. The CARD domain mediates the interaction between CARD-containing caspases and their upstream activators. Therefore, the scaffold molecule ASC, which is composed of an N-terminal pyrin domain (PYD) and a C-terminal CARD motif13,14, is required for recruitment of caspase-1 to the NLRP110, NLRP315, and AIM216 inflammasomes.
Each inflammasome is named after its unique sensor protein that recognizes distinct pro-inflammatory stimuli (Figure 1B). Activators of this pathway are termed canonical stimuli. Inflammasomes serve as sensors for microbial components and tissue stress, and assemble to trigger a robust inflammatory response through activation of the inflammatory caspases17. Inflammasome assembly initiates caspase-1 activation to mediate maturation and secretion of its main substrates pro-IL-1β and pro-IL-18. This process occurs via a two-step mechanism. First, a priming stimulus upregulates the expression of certain inflammasome proteins and pro-IL-1β through activation of the NF-κB pathway. Second, an intracellular (canonical) stimulus induces inflammasome assembly and recruitment of procaspase-16,7.
Caspase-4 and caspase-5 are the human orthologs of murine caspase-1111. They are activated in an inflammasome-independent manner by intracellular lipopolysaccharide (LPS), a molecule found in the outer membrane of Gram-negative bacteria18,19,20, and by extracellular heme, a product of red blood cell hemolysis21. It has been proposed that LPS binds directly to the CARD motif of these proteins and induces their oligomerization20. Activation of caspase-4 or caspase-5 promotes IL-1β release by inducing an inflammatory form of cell death called pyroptosis through cleavage of the pore-forming protein gasdermin D (GSDMD)18,19. In addition, the efflux of potassium ions resulting from caspase-4 and GSDMD-mediated pyroptotic death induces activation of the NLRP3 inflammasome and subsequent activation of caspase-122,23. Therefore, caspase-4, -5, and -11 are considered intracellular sensors for LPS that are able to induce pyroptosis and caspase-1 activation in response to specific stimuli11,24.
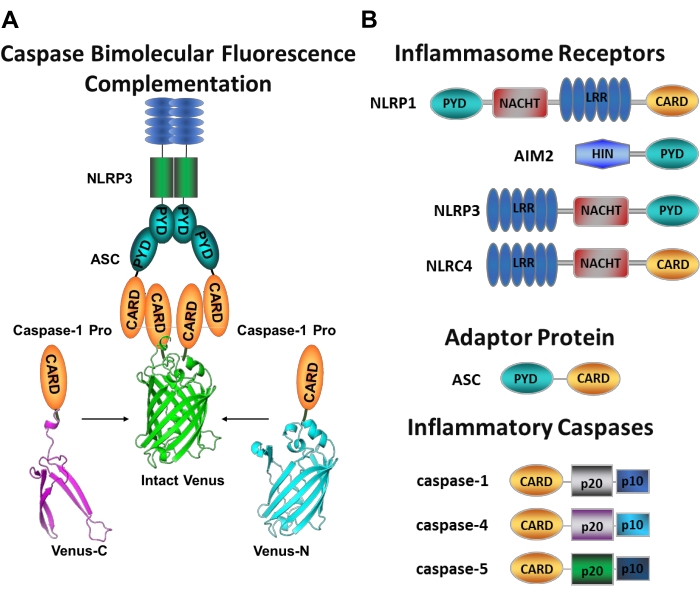
Figure 1: Inflammatory caspases and caspase-bimolecular fluorescence complementation (BiFC) assay. (A) Diagram showing the caspase-BiFC system, where two caspase-1 prodomains (C1-pro) linked to each non-fluorescent fragment of Venus (Venus-C or Venus-N) are recruited to the NLRP3 activation platform, forcing Venus to refold and fluoresce. This complex appears as a green spot under the microscope and serves as a readout for inflammatory caspase-induced proximity, which is the first step in initiator caspase activation. (B) Schematic showing the domain organization of inflammasome components and inflammatory caspases. Please click here to view a larger version of this figure.
Measuring specific initiator caspases activation is difficult, and there are not many methods available to do so by imaging approaches. Caspase Bimolecular Fluorescence Complementation (BiFC) can be used to visualize inflammatory caspase activation directly in living cells (Figure 1A)25. This technique has been recently adapted for use in human monocyte-derived macrophages (MDM)21. Caspase BiFC measures the first step in inflammatory caspase activation, induced proximity to facilitate dimerization. Expression of plasmids encoding the CARD-containing caspase prodomain fused to non-fluorescent fragments of the photostable yellow fluorescent protein Venus (Venus-C [VC]) and Venus-N [VN]) are used. When the two caspase prodomains are recruited to their activation platform or undergo induced proximity, the two halves of Venus are brought in close proximity and forced to refold and fluoresce (see Figure 1A,B). This provides a real-time readout of specific inflammatory caspase activation.
Human MDM abundantly express inflammasome genes and pattern recognition receptors that identify danger signals and pathogen products. This provides an ideal cell type for the interrogation of inflammatory caspase pathways. In addition, they can be derived from peripheral blood and even from patient samples to assess inflammatory caspase activation in a specific disease state. This protocol describes how to introduce the BiFC caspase reporters into MDM using nucleofection, an electroporation-based transfection method, how to treat the cells to induce inflammatory caspase activation, and how to visualize the active caspase complexes using microscopy approaches. Additionally, this methodology can be adapted to determine the molecular composition of these complexes, subcellular localization, kinetics, and size of these highly ordered structures25,26,27.
This protocol follows the guidelines of Baylor College of Medicine's human research ethics committee for the manipulation of human samples. Blood samples are handled following the institutional safety guidelines for human samples. Blood samples are obtained at a regional blood bank, where they are collected with citrate phosphate dextrose (CPD) solution. However, blood collected with other anticoagulants like sodium heparin, lithium heparin, or EDTA can also be used for this protocol28,29.
1. Isolation of human monocytes and differentiation into macrophages
- Obtain anticoagulated blood from de-identified healthy individuals at a regional blood bank and isolate peripheral blood mononuclear cells (PBMCs) as indicated below.
NOTE: Perform all steps in a tissue culture laminar flow hood. Use sterile tubes only and wear gloves. Add 10% bleach to all blood-related products when disposing. Sterile PBS (1x) or DPBS (without Ca2+ and Mg2+) can be used interchangeably.- Prepare the dilution buffer: Supplement 1x sterile PBS with 2% FBS and 0.5 mM EDTA.
- Prepare the culture medium: Supplement RPMI-1640 medium with FBS (10% (v/v)), glutamax (2 mM), and Penicillin/ Streptomycin (50 I.U./50 µg/mL)
- Precool the running buffer (Table of Materials) according to the manufacturer's protocol.
- Dilute whole blood with two volumes of dilution buffer. Using a serological pipet, transfer 15 mL of the anticoagulated blood to a 50 mL tube containing 30 mL of the dilution buffer. Mix gently by inversion.
- For each 10 mL of whole blood or 30 mL of diluted blood, add 15 mL of the density gradient medium to a 50 mL empty tube.
- Layer the density gradient medium from step 1.1.5 with 30 mL of diluted blood slowly and steadily using a 25 mL serological pipet. Keep the tip of the pipet against the wall of the tube and the tube at a tilted angle.
- Carefully transfer the tubes to a swinging-bucket centrifuge. Avoid disturbing the two phases. Centrifuge the tubes at 400 x g at room temperature (RT) for 25 min with acceleration and deceleration set to the minimum value.
- Carefully remove the top (clear) plasma layer using a 10 mL pipet and dispose of in a container with bleach (10%).
- Collect the interphase (white) layer of peripheral blood mononuclear cells (PBMCs, Figure 2) with a 10 mL pipet and transfer to a fresh 50 mL tube. Combine the white layer from different tubes of the same donor in a 50 mL tube up to 30 mL.
- Bring each tube to a total volume of 50 mL with the dilution buffer from step 1.1.1 and centrifuge at 300 x g and 4 °C for 10 min. Remove the supernatant with a 10 mL pipet and dispose of it in a container with bleach (10%).
- Resuspend each cell pellet in 1 mL of the pre-cooled running buffer from step 1.1.3 using a p1000 micropipette. Combine cell suspensions from the same donor in a new 15 mL tube. Bring the volume of each tube to 15 mL with pre-cooled running buffer and mix well by inversion.
- Take a 20 µL aliquot of the cell suspension from step 1.1.11 and prepare a 1:100 dilution using 1x sterile PBS. Determine the cell number using a hemocytometer.
- Centrifuge the cell suspension from step 1.1.11 at 300 x g and 4 °C for 10 min and remove the supernatant with a 10 mL pipet. If necessary, use a p200 micropipette to remove the supernatant completely.
- Resuspend the isolated PBMCs in 80 µL of pre-cooled MACS running buffer for each 1 x 107 cells, adding up to a maximum of 800 µL of the buffer.
- Add 20 µL of anti-human CD14 MicroBeads per each 1 x 107 cells or up to 100 µL per blood sample (~100 mL of undiluted blood). Mix well by inversion and place on a tube rotator for 20 min with continuous mixing at 4 °C.
- Remove the samples from the tube rotator, add 10 mL of pre-cooled running buffer to each tube, and centrifuge at 300 x g (acceleration = 5, deceleration = 5) and 4 °C for 10 min.
- Remove the supernatant with a 10 mL pipet and resuspend up to 1 x 108 cells in 500 µL of pre-cooled running buffer (2 x 108/mL).
- Perform the isolation of CD14-positive cells by magnetic cell sorting using a manual or automated system (Table of Materials) per the manufacturer's instructions.
- Take a 20 µL aliquot of the cell suspension from step 1.1.18 after CD14-positive selection and prepare a 1:100 dilution using 1x sterile PBS. Determine the cell number by counting the cells on a hemocytometer.
- Centrifuge the CD14-positive cells at 300 x g and RT for 10 min. Remove the supernatant using a 10 mL pipet or a vacuum system.
- Resuspend the cell pellet from step 1.1.20 in pre-warmed culture medium from step 1.1.2 to a final cell density of 1 x 107 cells/mL.
- Seed the isolated CD14-positive monocytes at a cell density of 5 x 106 cells.
- On a 10 cm tissue culture dish, add 10 mL of culture medium from step 1.1.2 supplemented with 50 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF).
- Add 0.5 mL of the cell suspension from step 1.1.21 to the culture medium dropwise and gently swirl the plate. Incubate cells in a humidified tissue culture incubator (37 °C, 5% CO2) overnight.
- The next day, aspirate the medium using a vacuum system to remove cells that did not attach overnight. Add 10 mL of fresh culture medium supplemented GM-CSF (50 ng/mL) and incubate cells in a humidified tissue culture incubator (37 °C, 5% CO2) for 7 days to allow complete differentiation (see Figure 3A for the appearance of CD14+ monocytes at various stages of differentiation in GM-CSF ). Exchange the culture medium every 2-3 days and supplement with fresh GM-CSF (50 ng/mL) each time.
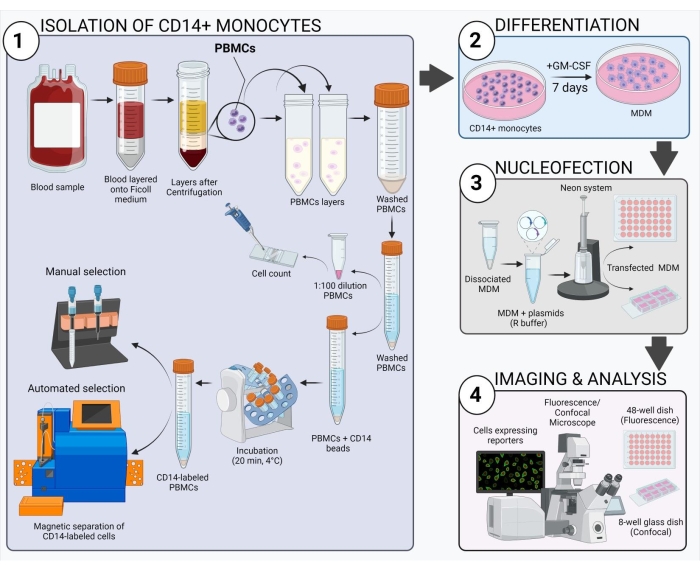
Figure 2: Schematic overview of the experimental workflow. Please click here to view a larger version of this figure.
2. Preparation of electroporation components
NOTE: This protocol is designed for a 10 µL-Neon tip (Table of Materials). For each transfection, use 1-2 x 105 cells. It is recommended to seed transfected cells on a 48-well plate or 8-well chambered dish (10 µL-transfected cells per well). 1x sterile DPBS (without Ca2+ and Mg2+) can be used in place of PBS.
- On day 7, prepare antibiotic-free medium by supplementing RPMI-1640 medium with FBS (10 % (v/v)) and glutamax (2 mM).
- Place serum-free RPMI-1640 medium, trypsin-EDTA (0.25%) solution, 1x sterile PBS (without Ca2+ and Mg2+), and complete culture medium from step 1.1.2 in a 37 °C water bath.
- If using glass-bottomed dishes (for confocal microscopy) coat the dishes with poly-D-lysine hydrobromide.
- Coat an 8 well-chambered dish with 200 µL of poly-D-lysine hydrobromide (0.1 mg/mL in 1x sterile PBS) and incubate for 5 min at RT.
- Aspirate the poly-D-lysine solution and wash the glass once with 1x sterile PBS. Aspirate the PBS and proceed with step 2.4.
- Add 200 µL of antibiotic-free medium per well of the 48 well plate or 8 well-chambered dish and pre-incubate in a humidified tissue culture incubator (37 °C, 5% CO2) until ready to plate the transfected cells.
3. Preparation of cells for electroporation
NOTE: The yield of MDM from a 10 cm dish at the end of the 7-day differentiation period is approximately 1.5 x 106 cells. 1x sterile DPBS (without Ca2+ and Mg2+) can be used in place of PBS. This protocol was optimized so that most macrophages are detached from the plate with the maintenance of cell viability and integrity. MDM are difficult to detach from cell culture plates. Therefore, it may be necessary to perform steps 3.2 and 3.3 twice to dissociate the cells. Ensure that each incubation time with trypsin-EDTA (0.25%) does not exceed 5 min.
- Aspirate the media from fully differentiated macrophages on 10 cm dishes and wash the cell monolayer with warm serum-free RPMI-1640 medium. Make sure to completely remove the medium.
- Harvest the cells by adding 2 mL of warm trypsin-EDTA (0.25%) solution per 10 cm dish and incubate in a humidified tissue culture incubator (37 °C, 5% CO2) for 5 min.
- Complete the cell detachment by gently pipetting the trypsin-EDTA (0.25%) solution up and down over the entire dish area using a p1000 micropipette. Transfer the cell suspension to a 15 mL conical tube containing 5 mL of warm complete culture medium from step 1.1.2.
- Take the dish to a bright field microscope and check for cell detachment in various fields of view. If there is a considerable amount of cells still attached, repeat steps 3.2-3.3.
- Centrifuge the cell suspension at 250 x g for 5 min at RT.
- Aspirate the medium and resuspend the cells in 10 mL of 1x sterile PBS pre-warmed to 37 °C. Take a 20 µL aliquot to determine the cell number using a hemocytometer.
- Take 1-2 x 105 cells per intended transfection and place in a 15 mL tube. Bring to a final volume of 15 mL with pre-warmed 1x sterile PBS. Centrifuge at 250 x g for 5 min at RT.
- Aspirate the PBS and centrifuge one more time for 1 min at 250 x g. Remove any residual PBS from the cell pellet using a p200 micropipette.
4. Nucleofection of caspase BiFC components into human monocyte-derived macrophages
NOTE: This section of the protocol is performed using the Neon Transfection System (Table of Materials). This protocol outlines the steps to transfect 1-2 x 105 cells using a 10 µL Neon tip (Table of Materials). If using a 100 µL Neon tip, scale up accordingly. Avoid exposing cells to resuspension buffer R for more than 15 min, as this may decrease cell viability and transfection efficiency.
- Dilute the reporter plasmid (i.e., mCherry or dsRedmito at 100 ng/µL) in nuclease-free water or 0.5x TE buffer to visualize the transfected cells.
- Dilute the caspase BiFC plasmids to an appropriate concentration in nuclease-free water or 0.5x TE buffer, so that the total volume of plasmids does not exceed 30% of the total transfection volume of 10 µL (i.e., 300 ng/µL of C1 Pro-VC and 300 ng/µL of C1 Pro-VN).
- Prepare a 1.5 mL sterile microtube per intended transfection and add the appropriate amount of reporter plasmid (i.e., 50 ng or 0.5 µL) and caspase BiFC plasmids (i.e., 300 ng or 1 µL of C1 Pro-VC and 300 ng or 1 µL of C1 Pro-VN fragment). Keep the microtubes in the hood at all times.
- Place the pipette station, device, tips, electroporation tubes, and pipette in a sterile laminar flow hood.
NOTE: The pipette station, device, tips, electroporation tubes, and pipette are included in the Neon Transfection System. - Connect the high voltage and sensor connector on the pipette station to the rear ports on the device as per the manufacturer's instructions. Keep the pipette station close to the device.
- Connect the power cord to the rear AC inlet and proceed to connect the device to the electrical outlet. Press the power switch to turn on the device.
- Enter the transfection parameters in the startup screen displayed when the device is switched on and appropriately connected. Press on Voltage, enter 1000, and press on Done to set the voltage to 1000 V. Press on Largura, enter 40, and press on Done to set the pulse duration to 40 ms. Lastly, press on # Pulses, enter 2, and press on Done to set the number of electrical pulses to 2.
- Take one of the electroporation tubes (provided in the kit) and fill it with 3 mL of electrolytic buffer E (for 10 µL tips and provided in the kit) at RT. Insert the electroporation tube into the pipette holder on the pipette station. Ensure that the electrode on the side of the tube is facing inwards and that a click sound is heard when the tube is inserted.
- Take the cell pellet from step 3.8 and add 10 µL of pre-warmed resuspension R buffer (provided in the kit) for each 1-2 x 105 cells. Mix gently with a p20 micropipette. Add 10 µL of the cell suspension to each tube set up in step 4.3 and mix gently with a p20 micropipette.
- Take the pipette and insert a tip by pressing the Push button to the second stop. Make sure the clamp completely picks up the mounting stem of the piston in the tip and that no gap is observed in the top-head of the pipette.
- To aspirate the sample, press the Push button on the pipette to the first stop and dip into the first tube containing cell/plasmid DNA mixture. Slowly aspirate the mixture into the pipette tip.
NOTE: Avoid air bubbles as they can cause arcing during electroporation, and if detected by the device, can prevent delivery of the electric pulse. If air bubbles are observed, release the contents into the tube and try aspirating again. - Insert the pipette with the sample very carefully into the pipette holder. Make sure the pipette clicks and that it is properly placed.
- Press Start on the touchscreen and wait until the electric pulses are delivered. A message on the screen will indicate completion.
- Slowly remove the pipette from the station and immediately add the transfected cell suspension into the corresponding well with pre-warmed antibiotic-free medium from step 2.4 by slowly pressing the push-button to the first stop.
NOTE: This tip can be reused up to three times for the same plasmid; otherwise, discard it into a biohazard waste container by pressing the Push button to the second stop. - Repeat steps 4.10-4.14 for each tube containing a cell/plasmid DNA mixture.
- Gently rock the plate with transfected cells and incubate for 1-3 h in a humidified tissue culture incubator (37 °C, 5% CO2).
- Add to each well 200 µL of pre-warmed culture medium (complete medium) from step 1.1.2. Place the dish in the humidified tissue culture incubator (37 °C, 5% CO2) again. Allow at least 24 h for gene expression.
- The next day, inspect the cell viability and transfection efficiency using an epifluorescence microscope.
- Turn on the epifluorescence microscope and the fluorescent light source box per the manufacturer's instructions and place the culture dish on the microscope stage.
- Select the 10x or 20x objective and the 568 nm (RFP) filter.
- To estimate cell viability, press the Transmitted Light LED (TL) button to visualize all cells in the selected field. While looking into the microscope eyepiece, turn the focus knob until cells are observed and check for cell attachment in the selected field.
NOTE: Fully attached cells represent the viable cells while floating cells represent non-viable cells. If the confluency of the well is high, the presence of non-attached cells could be the result of an overestimation of cell number and not the result of low viability. However, low confluency accompanied by a high content of floating cells signifies low viability that may result from arcing during electroporation, plasmid toxicity, or overexposure to resuspension R buffer. Do not use wells that display the latter behavior. - To estimate transfection efficiency, focus on cells in the selected field under transmitted light as described above. Count the total number of cells in the selected field. With the Transmitted Light LED (TL) switched off, press the Reflected Light LED button (RL) to switch on.
- Fine focus on the reporter gene fluorescence (red cells) and count the total number of red fluorescent cells. Repeat these steps (4.18.4-4.18.5) for at least two more fields per well.
5. Treatment of transfected MDM and caspase BiFC data acquisition
NOTE: If planning to image the cells using an epifluorescence or confocal microscope, treatment with qVD-OPh (20 µM) for 1 h prior treatment with the chosen stimulus is advised to prevent caspase-dependent cell death (predominantly apoptosis). This is used in imaging to prevent cells from lifting off due to apoptosis, making them very difficult to image as they move out of the focal plane. Note that caspase recruitment to the activation platform and the associated caspase BiFC is not dependent on the catalytic activity of the caspase, and consequently, caspase inhibition will not affect this step.
- Treat with chosen stimulus approximately 24 h after transfection and incubate for as long as necessary for each drug.
- Prepare imaging medium by supplementing culture medium from step 1.1.2 with Hepes (20 mM, pH 7.2-7.5) and 2-mercaptoethanol (55 µM).
- Add the desired concentration of stimulus to the pre-warmed imaging medium and mix gently.
- Remove the media from the cells carefully with a p1000 micropipette and add 500 µL of the stimulus solution from step 5.1.2 down the side of the well.
- To run untreated control wells, add imaging medium without the stimulus.
- Incubate the cells in a humidified tissue culture incubator (37 °C, 5% CO2) for as long as indicated for each treatment.
- Visualize the cells using an epifluorescence or confocal microscope.
- Turn on the microscope and the fluorescent light source, following the manufacturer's instructions.
- Select the 10x or 20x objective and place the culture dish on the microscope stage.
- Using the microscope eyepiece, find cells under the 568 nm filter and focus on the cells expressing the dsRedmito/mCherry reporter (red cells).
- Count all the red cells in the visual field and record the number.
- While in the same visual field, change to the 488 or 512 filter (GFP or YFP), proceed to count the number of red cells that are also green (Venus-positive or BiFC-positive) and record the number.
- Count at least 100 dsRedmito/mCherry -positive cells from a minimum of three individual visual fields.
- Calculate the percentage of Venus-positive transfected cells per visual field and average the resulting percentages for each treatment (well) to get the standard deviation.
- Image the cells using an epifluorescence or confocal microscope
NOTE: To acquire confocal images using a 20x objective or a greater magnification, cells should be plated on glass dishes unless the microscope is equipped with a long pass objective.- Follow steps 5.2.1-5.2.3. If using a confocal microscope with the 40x, 60x or 63x oil objective, place a drop of oil on the objective.
- Visualize the live image of the cells on the computer screen as acquired by the camera. Use the epifluorescence light source for fluorescent images or switch the light source to the lasers for confocal images.
- Fine-tune the focus and position of the cells using the joystick control and focus wheel.
- Set the percentage laser power and exposure time for the 512 nm or 488 nm (YFP or GFP) and 568 nm (RFP) lasers so that the signal in the image looks good and does not reach saturation.
- Turn on the live capture and examine the resulting image. Ensure that a distinct peak is seen for each fluor in the display histograms for both channels.
- Adjust the laser power and exposure time as necessary. Keep these values as low as possible while still being able to detect both fluorescent signals (RFP and GFP/YFP).
- While visualizing the live image of the cells, take multiple representative images of a field that contains one or more cells expressing the mCherry/dsRedmito reporter for each well of the plate and save the data.
The scheme shown in Figure 2 gives an overview of how to obtain, transfect, and image human MDM. After incubation of the selected CD14+ monocytes with GM-CSF for 7 days, the cell morphology changes over the course of the differentiation period (Figure 3A), going from spherical suspension cells to spindly and fully attached (days 3 and 4), and lastly to more spread cells when fully differentiated (day 7). Fully differentiated cells are then detached from the plate and transfected with the caspase BiFC pairs (VC and VN) along with the reporter plasmid (e.g., dsRedmito, a plasmid encoding a red fluorescent protein targeted to the mitochondria), which is used to label the transfected cells (Figure 3B) and employed to assess the efficiency of the transfection after 24 h. Figures 3B–D show an example of BiFC results using the caspase-1 pro BiFC transfected cells treated for 20 h with nigericin (5 µM), a known pro-inflammatory stimulus that triggers the assembly of the NLRP3 inflammasome30,31. Untreated cells show no Venus fluorescence (Figure 3B), and in nigericin-treated cells, caspase-1 BiFC appears as a single punctum with the typical shape of ASC specks32,33,25 (Figure 3C). Figure 3D shows an example of quantitation of Venus-positive MDM. The highest percentage of caspase-1 BiFC is seen in the LPS + nigericin treatment group (Figure 3D). This result is consistent with activation of a canonical inflammasome, where both a priming signal (LPS) and an intracellular signal (nigericin) are required for activation of capase-1.
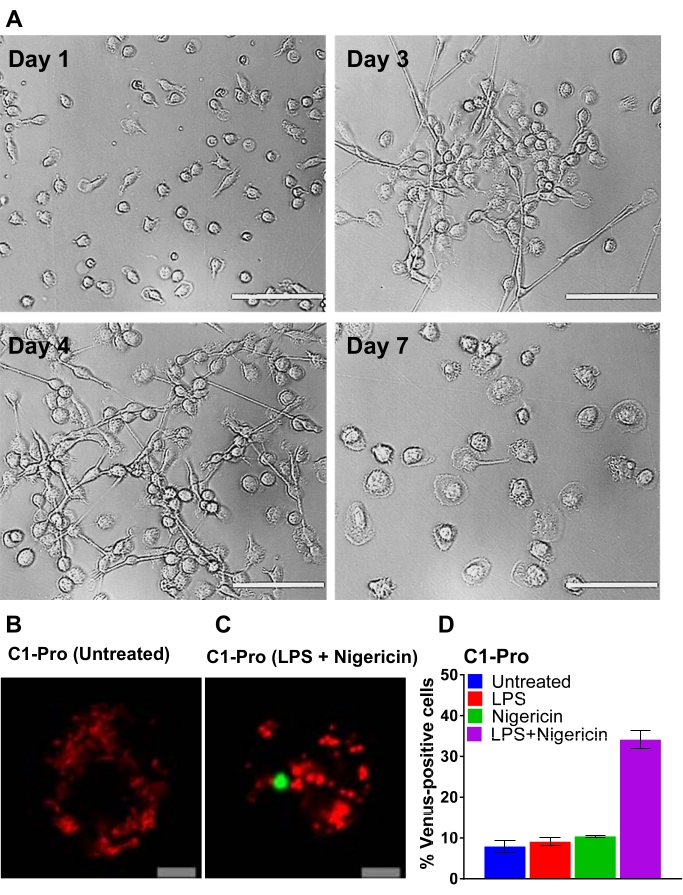
Figure 3: Differentiation of CD14-monocytes and transfection of human MDM. (A) Representative bright-field images of CD14+ monocytes from peripheral blood exposed to GM-CSF at days 1, 3, 4, and 7 of differentiation (Scale bar, 100 µm). (B–C) Human MDM were transfected with caspase-1 pro BiFC pairs and the transfection reporter dsRedmito (50 ng, red). 24 h later, cells were primed with and without LPS (100 ng/mL) for 3 h and treated with nigericin (5 µM) in imaging medium for 20 h. Representative confocal images of untreated and nigericin-treated cells are shown (Scale bar, 10 µm). The BiFC is shown in green. (D) Cells from (C) were assessed for the percentage of dsRedmito-positive cells (red, transfected cells) that were Venus-positive (green, caspase-1 BiFC complex) at 20 h. Error bars represent SD of at least two independent counts per well. Please click here to view a larger version of this figure.
An example of plasmid titration is shown in Figure 4, where increasing amounts of each BiFC plasmid is transfected. This allows for the selection of the optimal dose of plasmid, resulting in a specific signal and minimal background. Figure 4A shows the results of human MDM transfected with increasing concentrations of the caspase-1, -4, and -5 pro BiFC pairs (VC and VN) treated with heme, a highly pro-inflammatory molecule that results from red blood cell destruction34. The highest percentage of Venus-positive cells for caspase-1, -4, and -5 pro BiFC pairs results from 400 ng, 500 ng, and 1000 ng of transfected plasmid, respectively. However, using the highest amount of plasmid also runs the risk of increasing non-specific background (Figure 4B). For example, transfection of 1000 ng of the caspase-5 pro BiFC pair results in almost 40% non-specific background. Therefore, using the lower 700 ng amount is considered optimal for this plasmid pair (Figure 4B). In Figure 4C, representative confocal images obtained with a 20x objective of a field of cells 24 h post-transfection are shown. In this image, one can see that the untreated transfected cells are viable based on the appearance of the mitochondria (mitochondria in dead cells are highly fragmented) and the morphology of the cells (apoptotic cells are shrunken). After treatment with heme, the caspase-1 complex appears as a single green punctum similar to the one induced by nigericin in Figure 3C, and its appearance was accompanied by cell shrinkage.
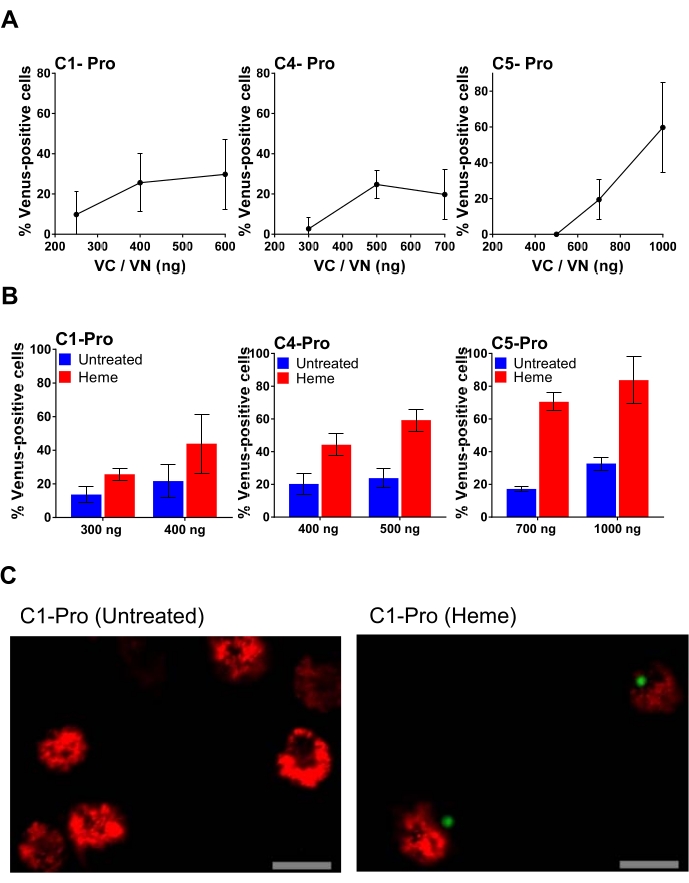
Figure 4: Human MDM transfected with different amounts of inflammatory caspase pro BiFC pairs. (A) Titration of caspase-1 pro; caspase-4 pro; or caspase-5 pro BiFC pairs. Human MDM were transfected with the indicated amounts of the caspase pro BiFC pairs with dsRedmito as a transfection reporter (50 ng) and incubated overnight. On the next day, the culture medium was removed from all wells, and cells were treated with and without heme (50 µM) in 0.1% FBS medium. After 1 h, an equal amount of complete medium was added to all wells to reconstitute the FBS concentration to 5% and prevent heme from killing the cells. After a total treatment of 20 h, cells were assessed for the percentage of dsRedmito-positive transfected cells that were Venus-positive, determined from a minimum of 300 cells per well. Error bars represent SD of at least three independent counts per well. (B) Human MDM were transfected with selected amounts of caspase-1 pro; caspase-4 pro; or caspase-5 pro BiFC pairs, along with dsRedmito (50 ng) as a reporter for transfection. 24 h post-transfection, cells were treated with or without heme (50 µM) in 0.1% FBS. After 1 h, FBS was reconstituted to 5% to inhibit extracellular heme. Cells were assessed for the percentage of dsRedmito-positive cells (red, transfected cells) that were Venus-positive (green, caspase BiFC complex) at 20 h determined from a minimum of 300 cells per well. Results are represented as percentage Venus-positive cells. Error bars represent SD of at least three independent counts per well. (C) Representative confocal images obtained with a 20x objective of a field of cells 24 h post-transfection and treated with and without heme as described in (B). Red: dsRedmito-positive cells; Green: Venus-positive cells; Scale bar, 20 µm. Please click here to view a larger version of this figure.
