Cell-generated mechanical forces are essential to proper function in various organs throughout the body such as the intestines, bladder, heart, and others. These organs must generate stable patterns of cell contraction and relaxation to maintain the internal homeostatic state. Abnormal smooth muscle cell (SMC) contraction can lead to the onset of various disorders, including, for example, intestinal dysmotility, characterized by abnormal patterns of intestinal smooth muscle contraction1, as well as the urologic conditions of overactive2 or underactive bladder3. Within the airways, SMCs that exhibit irregular contraction patterns can trigger asthmatic hyperresponsiveness4, potentially tightening the airways and decreasing airflow of oxygen into the lungs. Another widespread physical condition, hypertension, is caused by fluctuations in the smooth muscle contraction within blood vessels5. Clearly, contractile mechanisms within cells and tissues can lead to diseases that require treatment options. As these conditions unmistakably stem from the dysfunctional contractile behaviors of cells, it becomes logical and necessary to measure the cell contractile function itself, when screening potential drug candidates.
Recognizing the need for tools to study cellular contractile force, several quantitative contraction assay methods have been developed by academic researchers including traction force microscopy (TFM)6, micropatterned TFM7, floating gel assays8, and elastomeric micropost assays9. These technologies have been utilized in single-dish format as well as multi-well-plate format in numerous studies and have even been proposed for three-dimensional force measurements10,11,12,13,14. While these technologies have enabled pioneering research within the expansive field of cell force biology, they have all been largely limited to labs possessing specific capabilities and resources, in particular: ability to fabricate TFM substrates, ability to properly apply complex and non-intuitive algorithms to solve TFM displacement maps, and relatively precise microscopy systems that can register images taken before and after sample removal from the stage (for cell dissociation). Thus, for an untrained researcher, the entry barrier to use these methods can be quite high given the extensive set of requirements to apply these technologies. In addition, the imaging resolution required for many existing technologies (40x objectives or greater) can significantly limit experimental throughput, while bulk measurement technologies could mask contributions from outlier cells and prevent discovery of milder contractile differences. Of note, as far as the authors are aware, only the low-throughput and semi-quantitative floating gel assay approach has matured sufficiently to become available to researchers (see Figure 1).
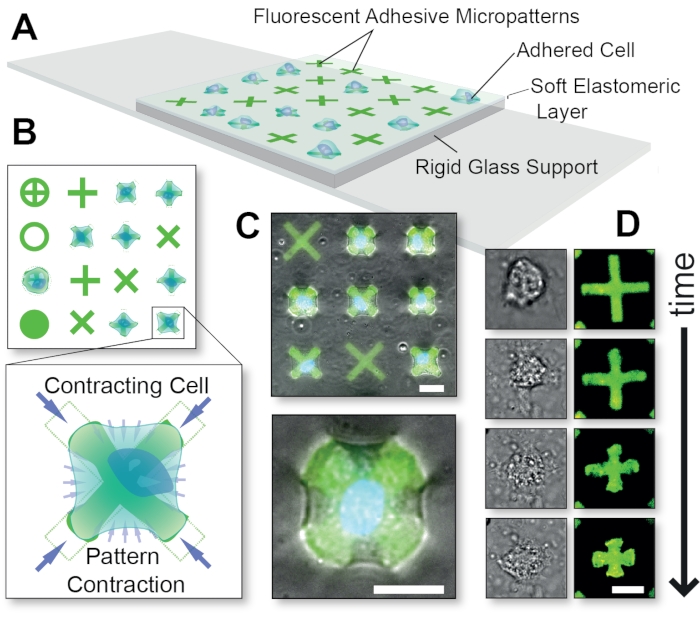
Figure 1: Overall schematic of the FLECS Technology method. (A) Cells are adhered to adhesive protein micropatterns that are covalently embedded into a thin elastomeric layer supported by glass. (B) Top-view of various possible micropattern shapes and a blow-up of a cell contracting an 'X'-shaped micropattern. (C) Overlay of fluorescent micropatterns and phase contrast images of a contracting cell. (D) Time-course images of a single contracting cell. Scale bars = 25 µm. This figure was adapted with permission from Pushkarsky et al15. Please click here to view a larger version of this figure.
Following recent advances in microtechnology, the authors developed a microplate-based technology enabling quantitative measurements of single-cell contraction in hundreds of thousands of cells called FLECS (fluorescently labelled elastomeric contractible surfaces)15,16,17,18,19,20, as an alternative to TFM. In this approach, fluorescent protein micropatterns are embedded into soft films which deform and shrink when cells apply traction forces to them, in an intuitive and measurable manner. Importantly, the protein micropatterns constrain cell position, shape, and spread area, leading to uniform test conditions. These allow simple measurements based only on their dimensional changes, which are highly resolved spatially even in 4x magnification images. The method includes a browser-based image analysis module and enables straightforward analysis of contractile cell force without requiring delicate handling procedures or registration of fiduciary markers, such that it should be operable by any researcher with a basic cell culture facility and simple fluorescent microscope with low magnification (Figure 2). This technology, which is shelf-ready and commercially available, was designed with the end-user in mind and aims to reduce the entry barrier for any laboratory scientist to study cellular force biology.

Figure 2: Schematic of the 24-well plate format for the single-cell contractility assay. This format was used in the experiments described herein and depicted in the video portion of the article. Scale bars = 25 µm. Please click here to view a larger version of this figure.
In this work, we present a protocol for applying the 24 well plate format of the FLECS Technology platform to quantify the effects of force-modulating drugs on cellular contractility in primary bladder smooth muscle cells. This general-purpose protocol can be adapted and modified as needed to account for various other timescales, cell-types, and treatment conditions of interest, and scaled to answer other questions in force biology.
Regions of the images acquired from wells that were treated with DMSO only and those that were treated with 40 µM blebbistatin are shown side-by-side in Figure 3. It can be clearly observed that DMSO-only treated cells exhibit a significant level of contraction based on the very prominent deformations of the micropatterns adhered to by bladder smooth muscle cells (BSMCs) in that well. Conversely, in the image of the well treated with 40 µM blebbistatin, significant cell relaxation is observed7 as the micropatterns adhered to by BSMCs are nearly indistinguishable in size from the micropatterns that are not adhered to by cells, indicating minimal contraction. These images demonstrate the intuitive and clear visual representation of single-cell contractility offered by the fluorescent micropatterning method. Unlike TFM-based methods, where the omnidirectional movement of numerous fluorescent particles randomly distributed beneath a dense cell monolayer is meant to convey relative contractile force, here, the uniform and marked contracted geometries of the micropatterns provide immediate and easily interpretable qualitative information about contraction of individual cells. These can be directly quantified by applying standard binary object operations on the images.
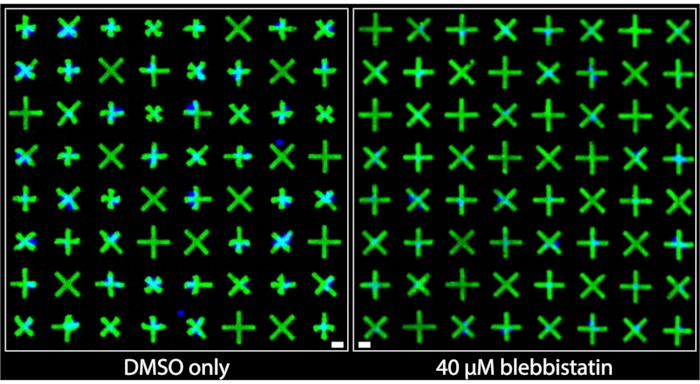
Figure 3: Side-by-side comparisons of images taken of wells containing either only 1% DMSO treatment (left) or containing 40 µM of blebbistatin (right). It can be clearly observed that treatment with blebbistatin significantly reduces the contractility of the single cells as indicated by the larger, un-contracted micropatterns. Blue nuclei indicate which micropatterns are bound by cells. Scale bar = 25 µm. Please click here to view a larger version of this figure.
By applying the browser-based analysis module to analyze the acquired image pairs of micropatterns and cell nuclei, single-cell contractility distributions are obtained for each population as shown in Figure 4. Described in detail in a prior report on the FLECS methodology15, the analysis works by locating the positions and orientations of each "X" shaped micropattern, counting the number of nuclei adhered directly over the center of each micropattern, computing the mean length of each micropattern, and calculating the pixel distance of the contraction of each micropattern with respect to the mean length of empty micropatterns (zero contraction reference). Therefore, the empty micropatterns serve an important purpose for normalizing the contraction data. Importantly, cells that do not bind to the micropatterns will accumulate on the well boundaries due to microcurrents where they will not affect image analysis. As seen in these plots, the unperturbed contractility of the cell population treated only with DMSO controls spans a large range as high as 20 pixels, with a center found at around 10 pixels. Meanwhile, cells treated with blebbistatin contract significantly less and their distribution is pushed down to a center of just over 6 pixels. Importantly, every single micropattern found in the image that binds exactly on cell is represented in these distributions. This demonstrates the ability of the method to convey differential cellular responses to drug treatments.
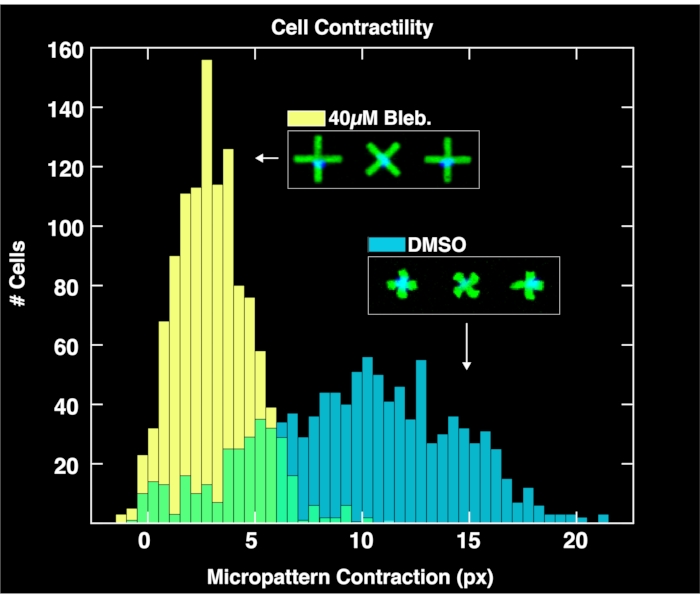
Figure 4: Histograms depicting single-cell contractility data obtained from the analysis of images taken of wells containing 1% DMSO only (blue) or 40µM blebbistatin. The distribution of DMSO-treated cells is broad and centered at a much larger contraction value (~10 pixels) than the blebbistatin treated distribution, demonstrating the quantitative effects of treatment of cells with blebbistatin. Please click here to view a larger version of this figure.
By utilizing all of the wells on a single 24-well plate and imaging at least 3 sites per well, six-point dose-response curves are simultaneously generated for two drug compounds. Figure 5 shows the concentration-response data for BSMCs treated with the same range of doses of blebbistatin or cytochalasin D (both known contractility inhibitors). As evident from the concentration-response profiles, cytochalasin D is the more potent inhibitor of tonic contraction in these cells. By fitting a sigmoidal curve to the data points, IC50 values can be calculated for each drug. Our experiments indicate that the IC50 are 7.9 µM and 100 nM for blebbistatin and cytochalasin D, respectively, following ~30 min of exposure to the drugs. Importantly, overall, these values are consistent with prior reports, validating the quantitative accuracy of the method to determine potency of contraction inhibitors7,21.
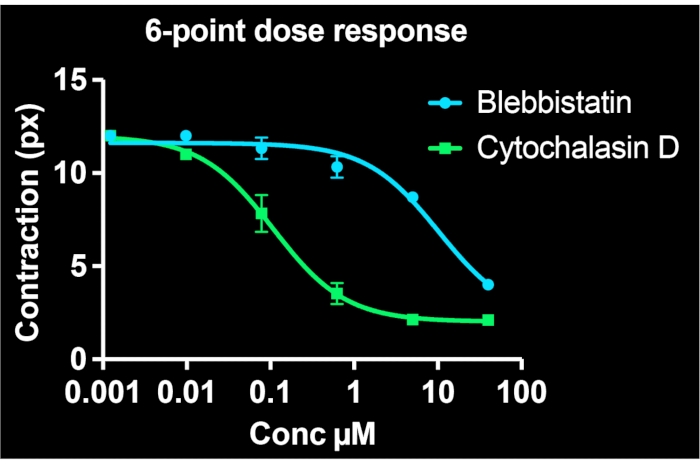
Figure 5: Concentration-response curves depicting the effects of blebbistatin and cytochalasin D on cellular contractility in single-cells. Each data point comprises three images for that condition. A sigmoidal curve was fit to each set of data. The results indicate that cytochalasin D is more potent, having a lower IC50 value. This data is collectible from a single 24-well FLECS plate. Please click here to view a larger version of this figure.
