This protocol has been used to collect Caenorhabditis nematodes from multiple locations, including Hawaii and California. The isolation success rate for Caenorhabditis nematodes varies with collection location, climate, sampling experience, and substrate types sampled. The protocol has been used to extensively sample the Hawaiian Islands, where nine collection projects have been conducted over multiple years and seasons. The isolation success rates for selfing Caenorhabditis species are nearly identical for C. briggsae (162 of 4,506 samples, 3.6%) and C. elegans (163 of 4,506 samples, 3.6%), and much lower for C. tropicalis (26 of 4,506 samples, 0.58%)8. Each of the selfing species is enriched on rotting fruit and flower substrates relative to the other substrate categories. Sample rotting fruit and flower substrates if the researcher is attempting to maximize the success rate rather than characterize substrate preferences. However, the success rate varies with the quality of the substrate selected. For example, among fruit and flower substrates, those substrates that are too dry, wet, or fresh will likely not yield Caenorhabditis nematodes.
The scalability of this collection protocol is evident from the number of collections a single pair of researchers can collect from the wild. For example, in October of 2018, a pair of researchers using this collection protocol was able to collect a total of over 1,000 samples in 7 days from multiple locations on two Hawaiian Islands. This field team shipped the samples overnight to the laboratory, where a team of eight researchers isolated over 2,000 nematodes from the samples as they arrived. A key advantage of this protocol is that it minimizes the cost associated with sampling in remote locations by reducing the equipment and personnel required in the field. Using this protocol, a small field team can focus on sampling while the isolation team can process the samples at their home institution using fragile and heavy equipment like dissecting microscopes and agar plates for isolating nematodes. Moreover, the implementation of the mobile data-collection application allows all the field data associated with the samples to be linked directly to the C-label, which enables the isolation team to work independently from the field team while processing samples.
Researchers that use this collection protocol must consider the effort that is required to isolate nematodes prior to a collection project. The isolation and identification steps are rate-limiting, and a small collection team can quickly overwhelm isolators with samples. Moreover, the laboratory space required to process many collections can interfere with ongoing research (Figure 3). Additionally, some isolated nematodes require additional effort to genotype. For example, approximately 2% of isolates fail to amplify with the SSU PCR primer set after the first lysis attempt and must be re-lysed to ensure that the lysis material is suitable for amplification with the ITS2 primer set (Figure 8). Furthermore, approximately 3% of isolates fail to produce quality sequences after an initial round of Sanger sequencing. For these isolates, another round of lysis, ITS2 PCR, and Sanger sequencing is often required, which can increase handing time for the isolation team. Importantly, sequence identity alone is not sufficient evidence to justify a new Caenorhabditis species (Figure 7). To properly justify raising an isolate as a new Caenorhabditis species, additional effort must be made to perform mating experiments and establish a typed specimen13. A formal morphological description of the typed specimen is also preferred but not required3. Together, these considerations suggest that researchers adopting this collection protocol will benefit from trial tests of the isolation and identification steps to ensure resources are properly allocated before a collection project begins. Importantly, even small collection projects can benefit from this protocol because the process is highly reproducible, and the data can easily be audited for quality control purposes across laboratory groups.

Figure 1: Substrate examples. (A) An ideal rotting fruit is shown in the center of the image (1), the fruit is almost unrecognizable. Less rotted fruit is shown nearby; avoid sampling freshly fallen fruits (2). (B) An ideally decomposed flower is shown at the top (3). Avoid sampling freshly fallen flowers (4). (C) The dark leaf litter under the top layer of dry leaves is ideal when sampling for selfing Caenorhabditis nematodes (5). Avoid sampling dry leaf litter (6). Please click here to view a larger version of this figure.
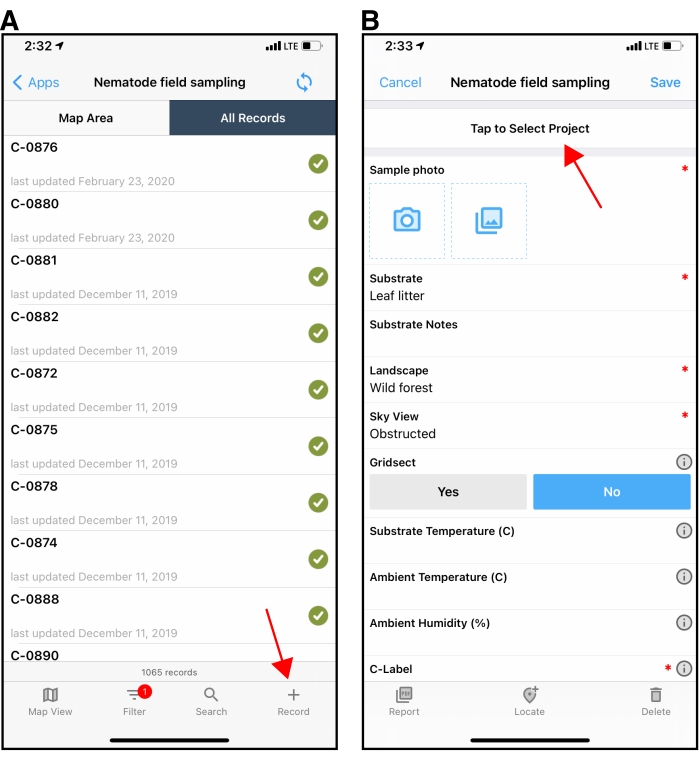
Figure 2: The Nematode Field Sampling mobile application. (A) The initial screen after opening the Nematode Field Sampling application on an Apple device in Fulcrum. The red arrow in the lower right points to the + button used to create a new collection record. (B) An example of a new collection record shown on an Apple device. The red arrow points to the 'Project' field at the top of the collection record screen. Be sure to select the correct project when sampling in the field. The project field will default to the last project used when creating subsequent collection records. Please click here to view a larger version of this figure.

Figure 3: Collection bags and collection plates organized prior to plating out samples. This figure shows the samples in C-labeled collection bags on the left. Each collection bag has a matching C-labeled 10 cm plate on top of it. On the right are 10 cm collection plates that contain sample material after it was transferred from the collection bags. Please click here to view a larger version of this figure.

Figure 4: A collection plate (C-plate) with properly a transferred sample. A 10 cm C-plate with decomposing fruit placed on the edge of the bacterial lawn. The C-label is attached to the plate lid. Please click here to view a larger version of this figure.
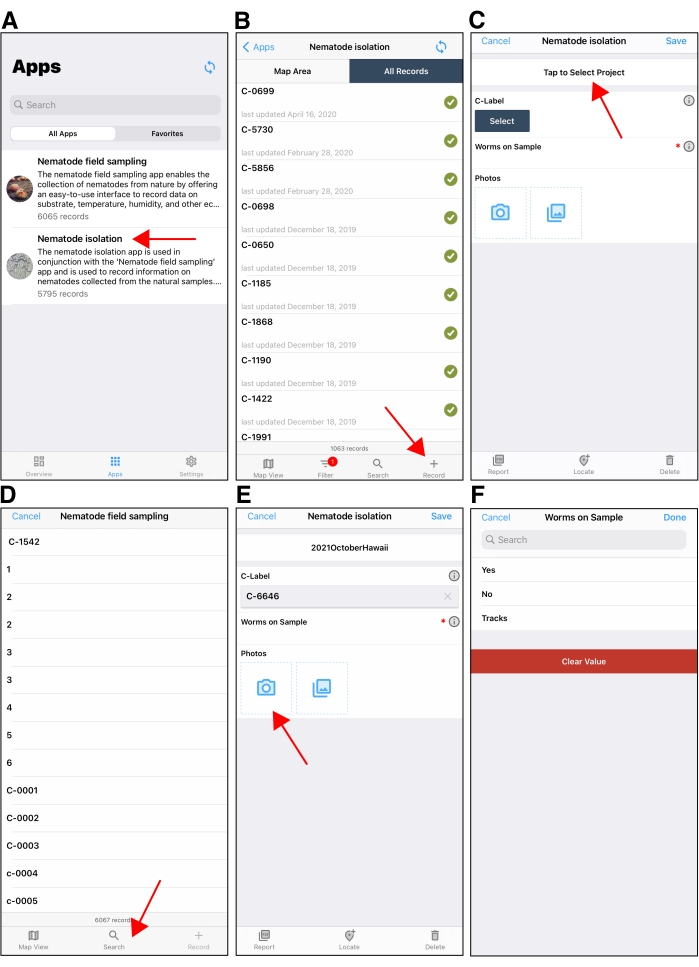
Figure 5: The Nematode isolation mobile application. (A) The application selection screen in the Fulcrum mobile application. The red arrow points to the Nematode Isolation application. (B) The initial screen after opening the Nematode Isolation application on an Apple device in Fulcrum. The red arrow in the lower right points to the + button used to create a new isolation record. (C) An example of a new isolation record shown on an Apple device. The red arrow points to the 'Project' field at the top of the isolation record screen. Be sure to select the correct project when isolating. The project field will default to the last project used when creating subsequent isolation records. (D) After tapping the Select field under C-label, users will tap the search button (red arrow) to find the C-label from which they are isolating nematodes. (E) After the C-label is selected, users will photograph the C-plate using the device camera. (F) Users then input whether there are nematodes on the C-plate or not. S-labels are added to the isolation record if there are nematodes to be isolated. Please click here to view a larger version of this figure.
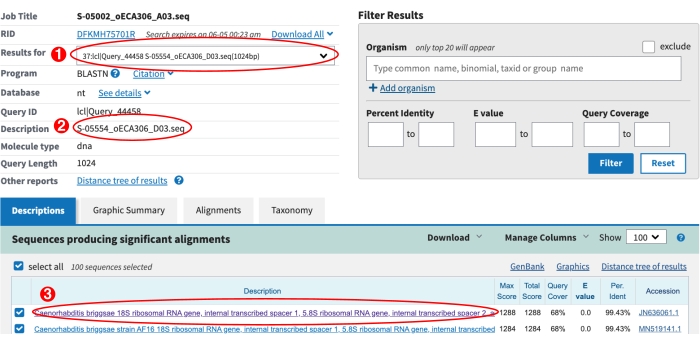
Figure 6: NCBI BLAST results page. (1) The drop-down menu used to view the BLAST results for all sequences. (2) The description of the current sequence selected from the drop-down. In this case the results for S-label S-05554 are shown. (3) The top BLAST hit for S-05554 is shown. The purple text indicates the link to visualize this alignment has been clicked. Please be sure to inspect the alignments by eye to identify possible new Caenorhabditis species, see step 9.8 above. Please click here to view a larger version of this figure.

Figure 7: NCBI BLAST alignment visualization examples. (A) An example of an isolate's ITS2 query sequence aligned to a C. kamaaina subject sequence. (1) The percent identity of the alignment (89%), which is low for a top BLAST hit. (2) A mismatch between the query and subject sequence (G to A). (3) A four base pair gap in the subject sequence made by the alignment algorithm; gaps in the query or subject indicate poor alignment. (4) A generalized region in the center of the alignment with many mismatches and gaps. A region like this one suggests that the query sequence might come from a new Caenorhabditis species. Shown is an actual alignment example of a new species, C. oiwi, that was discovered in 2017. (B) An example of a good alignment between an isolate's ITS2 query sequence and a subject sequence. (5) The percent identity of the alignment (99%), which usually means the query sequence comes from an isolate of the same species as the subject. (6) A central region of the alignment with perfect identity. A region like this one suggests that the query isolate is likely the same species as the subject. Please click here to view a larger version of this figure.
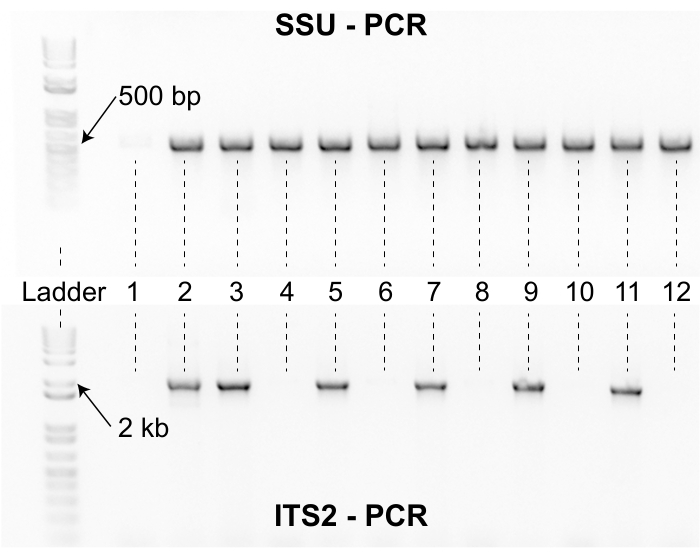
Figure 8: SSU and ITS2 PCR products. The top gel shows PCR products generated with the SSU primer set for 12 representative samples. A DNA ladder is included on the left as a reference. The SSU PCR products for Caenorhabditis nematodes are approximately 500 bp in length. Samples 2-12 amplified with the SSU primer set but sample one did not. The absence of a 500 bp SSU amplicon for sample one suggests that the lysis material was of poor quality and the sample must be re-lysed. The bottom gel shows PCR products generated with the ITS2 primer set for the same 12 Samples shown in the top gel. The ladder and samples are in the same orientation for both gels. Six of the 12 samples did not amplify with the ITS2 primer set. The samples with SSU and ITS2 bands are Sanger sequenced and identified by sequence similarity using the NCBI BLAST algorithm. Please click here to view a larger version of this figure.
Supplemental File 1: C-labels. A PDF file containing 2500 unique C-labels. Please click here to download this File.
Supplemental File 2: S-labels. A PDF file containing 5000 unique S-labels. Please click here to download this File.
Supplemental Table 1: Field Materials. A packing list of materials used in the field to sample nematodes. Please click here to download this Table.
Supplemental Table 2: PCR recipes and thermocycler conditions. A table of PCR recipes and thermocycler conditions for the ITS2 and SSU PCRs. Please click here to download this Table.
Supplemental Table 3: Electrophoresis buffer recipes. A recipe for 0.5 M pH 8.0 Ethylenediaminetetraacetic acid solution (EDTA) and the TRIS-acetate-EDTA (TAE) buffer solution. Please click here to download this Table.
