The sea anemones were collected according to guidelines of the National Commission for Aquaculture, Fisheries, and Food of the Federal Government of Mexico (permit number PPF / DGOPTA 07332.250810.4060). Bioethics Committee of the Institute of Biotechnology, National Autonomous University of Mexico approved all the experiments with sea anemones. The sheep blood sample was purchased at the Center for Practical Teaching and Research in Animal Production and Health (CEPIPSA, National Autonomous University of Mexico).
1. Organism collection
- Collect sea anemone A. dowii.
NOTE: Here, the organisms were collected during low tide periods in the intertidal zone off the coast of Ensenada Baja, California, Mexico (Figure 1A,B). - Transport the organisms to the laboratory in containers with seawater (Figure 1C).
- In the laboratory, clean the organisms with distilled water by hand to remove the big substrate particles adhering to the body.
- Immediately freeze the organisms at -80 °C in an ultra-freezer for 72 h.
- Place the organisms in special glasses for freeze-drying, with lyophilization conditions of -20 °C, 0.015 psi, for 48 h.
- Store the lyophilized organisms at -20 °C until use.
2. Tissue hydration
- Reconstitute 20 g dry weight of lyophilized organisms with 60 mL (1/3 w/v) of 50 mM sodium phosphate buffer, pH 7.5, and 1 inhibitor cocktail tablet (Table of Materials).
- In a magnetic stirrer, continuously stir the sample at ~800 rpm, 4 °C, for 12 h.
3. Toxin release
- To induce cell lysis and nematocyst discharge, freeze the sample at -20 °C for 12 h, and then place the container in water at room temperature (RT) until it thaws up to 90%. Repeat this procedure three times.
NOTE: The sample does not thaw 100%; it is crucial to maintain the sample at ~4 °C to prevent protein degradation. - Observe the nematocyst discharged under a confocal microscope. Take 10 µL of the sample on a slide and place a coverslip over it. Observe under a confocal microscope at 100x and 60x magnification. Here a dye was not necessary.
- Process the images captured in the confocal microscope in ImageJ software (version 1.53c, Wayne Rasband, National Institutes of Health, USA, https://imagej.nih.gov/ij).
- If the tubule inside the nematocyst uncoils and is outside, the nematocysts are discharged; then, continue with the next step. Otherwise, only conduct one more freeze-thaw cycle.
- To clarify the extract, centrifuge the sample at 25,400 x g for 40 min, 4 °C, recover the supernatant, and centrifuge it again. Repeat this step 3-4 times.
- Recover the supernatant and discard the pellet. The supernatant or crude extract contains the venom components (toxins) and other cell molecules, such as lipids, carbohydrates, and nucleic acids.
4. Quantitation of total protein
- Determine the protein concentration of the crude extract using a commercial Bradford colorimetric assay15 kit (Table of Materials).
- Dilute the dye reagent (one part of dye with four parts of distilled water).
- Prepare bovine serum albumin (BSA) Fraction V (Table of Materials) solution at 1 mg/mL concentration in phosphate buffer (50 mM sodium phosphate, pH 7.5).
- Prepare the solutions listed in Table 1 in triplicate.
- Vigorously mix each solution in a shaker for 4 s.
- Incubate the samples for 5 min at RT, without shaking.
- Measure absorbance (AU) at 595 nm.
- Plot the averages of the sample BSA absorbances (AU vs. protein concentration), and determine the equation of the graph (Equation 1).
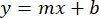 Equation 1
Equation 1
where y is the absorbance, m is the slope of the line, x is the protein concentration, and b is the y-intercept. To calculate crude extract concentration, solve for the x as follows:
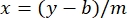 Equation 2
Equation 2
5. Determine polypeptide venom complexity
- Assemble the glass container for vertical gel electrophoresis according to the manual.
- Prepare acrylamide mix (30%): 29.2 g of acrylamide, 0.8 g of bis-acrylamide, the final volume of 100 mL. Filter the solution using a 0.45 µm membrane. Store the solution in a dark bottle at 4 °C. Then, prepare the mixture for the resolving gel (Table 2).
- The SDS-PAGE gel consists of a resolving gel and a stacking gel. Prepare the gels as per the solution volumes defined in Table 2. During preparation, ensure to maintain each mix at 4 °C to reduce polymerization time.
NOTE: The volume of each mixture varies depending on the brand of equipment used; for this protocol, the volumes correspond to the preparation of 15% acrylamide gel, 0.75 mm thick, for a vertical electrophoresis chamber (Table of Materials). - After the acrylamide has polymerized, ~10 min, add the mixture of stacking gel.
- Prepare the mixture for the stacking gel, immediately add it to the glass plates, and place a comb to form the wells for loading the samples.
- Once the stacking gel has polymerized (~15 min), remove the comb and place the glass container in a chamber for vertical electrophoresis.
- Place 200 mL of electrode buffer (0.25 M Tris, 0.192 M glycine, 0.1% SDS) in the electrophoresis chamber.
- Analyze the crude extract: Mix 30 µg of crude extract with 5 µL of protein loading buffer (25% glycerol, 15% sodium dodecyl sulfate, 25% 2-mercaptoethanol, 0.125 mM Tris pH 7, bromophenol blue 0.1 mg/mL) to denature proteins. The final volume must be <50 µL.
- Heat at 90 °C for 5 min, cool, and centrifuge for 3 s at 1,400 x g.
- Load the samples in the wells of the stacking gel. Load a standard molecular weight ladder.
- Run electrophoresis at 25 mA constant for 1 h 30 min.
- Remove the glass containers from the electrophoresis chamber to proceed with the staining of the gel proteins.
6. Protein staining
- Rinse the gel in 50 mL of distilled water to remove debris from the buffer electrode.
- Discard the water.
- To avoid the diffusion of the gel proteins, add the fixation solution (60 mL of ethanol, 20 mL of acetic acid, and 20 mL of distilled water, final volume 100 mL). Incubate at RT for 40 min with shaking at 80 rpm. Decant the solution.
- Add the protein crosslinking solution (15 mL of ethanol, 0.25 mL of glutaraldehyde, 50 mL of distilled water, final volume 65.25 mL), and incubate for 30 min at RT with shaking at 80 rpm. Remove the solution.
- Wash the gel with 100 mL of distilled water for 5 min, and remove the water. Repeat this procedure four times.
- Carry out protein staining with 50 mL of Coomassie brilliant blue R-250 filtered solution (40% methanol, 10% acetic acid, 50% distilled water, 0.1% dye, final volume 100 mL), stirring at 60 rpm in a laboratory rotary oscillator at RT for 15 min.
- Recover the solution, which can be used to stain other gels.
- To eliminate the excess dye, add a destaining solution (40 mL of methanol, 10 mL of acetic acid, 50 mL of distilled water). Keep stirring (80 rpm) at RT until the bands (stained proteins) visualize.
- Keep the gel in 50 mL of distilled water and scan the image in a Gel documentation system.
7. Prepare red blood cell solution
- Extract 1 mL of blood from the jugular vein of a sheep.
- Remove the needle from the syringe and immediately drain blood into a tube with 50 mL of Alsever solution (0.002 M citric acid, 0.07 M NaCl, 0.1 M dextrose, and 0.027 M sodium citrate as an anticoagulant, pH 7.4).
- Slowly invert the solution three times.
- Keep at 4 °C for transfer to the laboratory.
- Centrifuge at 804 x g for 5 min at 4 °C and discard the supernatant.
- Resuspend the pellet in 30 mL of Alsever solution. Add the solution by sliding it along the tube's inner walls and, using a Pasteur plastic pipette, slowly resuspend the cells. Centrifuge again at 804 x g for 5 min, 4 °C. Repeat this procedure until the supernatant is clarified.
- Add 15 mL of Alsever solution; resuspend the pellet slowly with a Pasteur plastic pipette.
- Maintain the final volume of Alsever solution to achieve a concentration of 2 x 106 cells/mL, approximately. Then, use a Neubauer chamber or hemocytometer to count the cells.
NOTE: Count the erythrocytes in the hemacytometer slide (Improved Neubauer). The slide is a 30 mm x 70 mm x 4 mm slide. The center has two silver footplates (Figure 2A) (one top and one bottom grid), each 3 mm x 3 mm, and two channels on the sides of the grid. Counting cells is in the corner and center squares (red squares in Figure 2B). The optimal concentration of cells for counting should be in the order of 106 cells. The central box has a surface area of 0.1 cm2, which is equivalent to 0.0001 mL. - Place a coverslip over the slide.
- Place 10 µL of the sample between the slide and the coverslip and wait for the sample to disburse.
- In an optical microscope (magnification 60x), perform the count in the first square box. Count only the cells in the center grid and those that touch the upper or the left margin of the box.
- Determine cell concentration using the following equation:
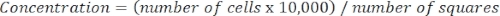 Equation 3
Equation 3
If the sample is very concentrated and dilution is required, then use the following equation:
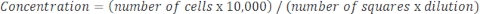 Equation 4
Equation 4
NOTE: When taking the erythrocyte solution for the hemolysis test, slowly homogenize the solution with a plastic Pasteur pipette. - Perform the steps in triplicate to reduce error.
8. Hemolysis assay
- Prepare the solution mixtures (in triplicates) indicated in Table 3 in conical 96-well plates.
NOTE: Mix the erythrocytes slowly to maintain a homogeneous suspension. - Incubate for 1 h at 37 °C, without shaking.
- Centrifuge the plate at 804 x g for 5 min at 4 °C.
- Recover the supernatant in another 96-well plate with a flat bottom.
- If the crude extract contains cytolysins, erythrocytes will lyse and release hemoglobin. Read the absorbance at 415 nm.
- Calculate hemolysis percentage, using the following equation:
% Hemolysis = ((Acrude extract-AAlsever buffer) / (AH2O-AAlsever)) x 100
NOTE: Acrude extract, AAlsever buffer, and AH2O correspond to the absorbance of erythrocytes with the crude extract, absorbance of Alsever solution, and absorbance of distilled water, respectively. - Perform a hemolysis test before and after each freezing and thawing cycle with 50 µg of total protein from the crude extract.
- Determine the number of freezing and thawing cycles required to reach 100% hemolytic activity.
- Calculate the amount of extract that produces hemolysis in 50% of the erythrocytes (HU50); follow the same protocol described in Table 3, increase the amount of sea anemone crude extract, and design the plot with sigmoidal adjustments using appropriate software (e.g., Origin, Table of Materials).
9. Phospholipase assay
- Wash one chicken egg with 1% SDS in distilled water.
- Separate the egg yolk from the egg white under sterile conditions.
- Prepare 50 mL of 0.86% NaCl solution, and filter through a 0.22 µm filter. Then, prepare Solution A: Add 12 mL of egg yolk and 36 mL of 0.86% NaCl solution.
- Prepare Solution B: Mix 300 mg of agarose in 50 mL of buffer (50 mM Tris, pH 7.5), filter through a 0.22 µm filter. Heat the solution in a microwave until boiling. Cool down in warm water to reach 43-45°C.
- Prepare Solution C: Prepare 10 mM CaCl2 and filter it with a 0.22 µm filter.
- Prepare Solution D: 10 mg of rhodamine 6G in 1 mL of distilled water.
- Under sterile conditions (laminar flow hood), add 500 µL of solutions A and C to solution B and 100 µL of solution D, mix, and pour 25 mL into each Petri dish (90 x 15 mm).
- Wait for the solution to solidify under sterile conditions (30 min).
- Make wells (~2-3 mm diameter) with a thin tube.
- Add a total of 20 µL of phosphate buffer (negative control) in one well and 20 µL of a determined phospholipase (positive control) in another well.
- Place different amounts of the crude extract protein, 5, 15, 25, 35, 45 µg, in the remaining wells, each in a final volume of 20 µL.
- Wait for the agar to adsorb all the samples (30 min).
- Incubate at 37 °C for 20 h.
- If a halo forms around the well, this indicates phospholipase activity. Measure the halo formed with a vernier caliper.
NOTE: Perform the experiment in triplicate (steps from 9.1-9.14).
The representative results of the protocol used to obtain the crude extract of sea anemone showed that combining two techniques (agitation and cycles of freezing and thawing) produced an efficient discharge of nematocysts, and the total amount of protein was 500 mg (8 mg/mL) (Figure 3).
The crude extract's protein complexity could be observed from 10 kDa and greater than 250 kDa through SDS-PAGE electrophoresis. In addition, cytolysins were detected in the molecular weight zone of 15 kDa and 20 kDa, a range of molecular weights that may correspond to phospholipases13 or actinoporins3 (Figure 4).
The hemolytic activity of the extract before and during the freezing and thawing cycles increased until 100% hemolysis was reached with 50 µg of the total crude extract protein in the last two cycles. The amount needed to lyse 50% sheep erythrocytes (HU50) is 11.1 ± 0.3 µg/mL (Figure 5). Phospholipase activity was detected by the formation of clear halos in the areas of the agar plate, where the crude extract sample was applied. The results show the presence of phospholipase activity from 15 µg of total protein. The diameter of the halo increased in a dose-dependent manner, i.e., if the amount of crude extract increased, the diameter of the halo increased (Figure 6A and Table 4).

Figure 1: Sea anemone collection. (A) Intertidal zone in El Sauzal, Baja California Norte, Mexico. (B) Sea anemone collected. (C) Anthoplerua dowii Verrill, 1869. Please click here to view a larger version of this figure.
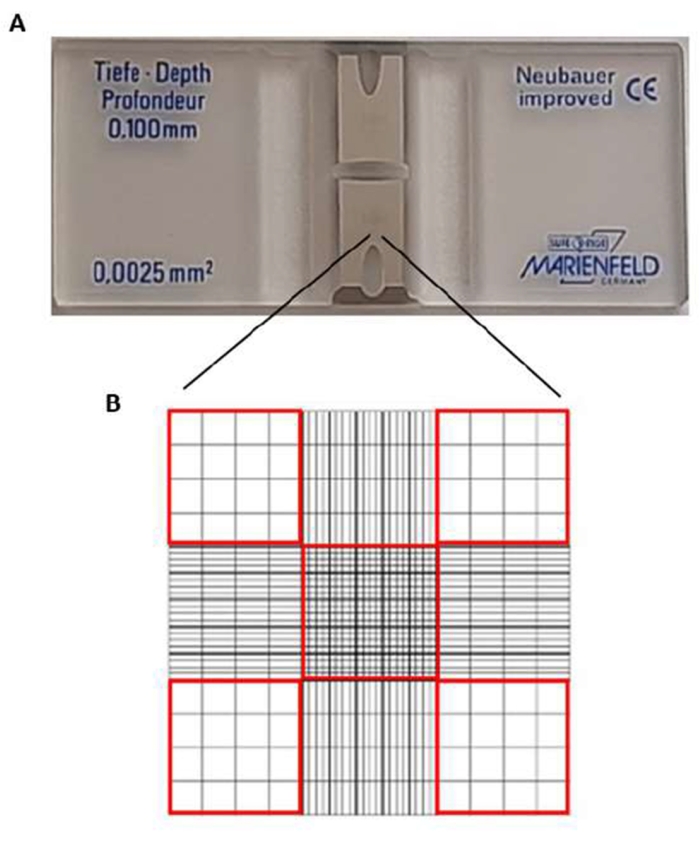
Figure 2: Hemacytometer slide. (A) In the slide center, there are two silver footplates. (B) Cells in squares with a red perimeter are counted. Please click here to view a larger version of this figure.
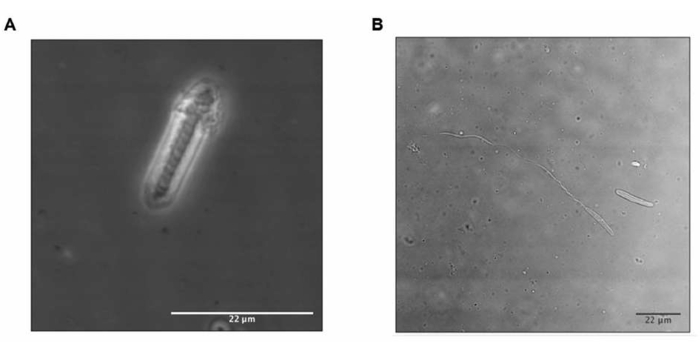
Figure 3: Nematocyst discharge. Nematocyst (A) before and (B) after stimuli. In B, the nematocyst tubule is exposed and indicates toxin release. Please click here to view a larger version of this figure.
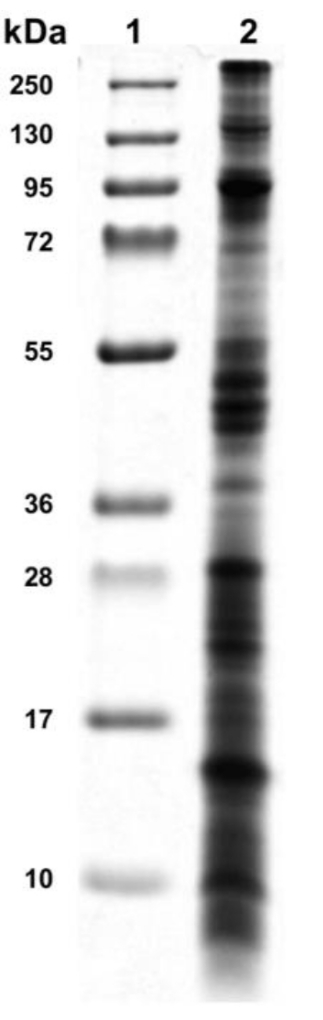
Figure 4: Electrophoretic profile of sea anemone crude extract. The sea anemone crude extract was analyzed by SDS-PAGE 15% polyacrylamide gel and showed protein from 10 kDa to 250 kDa of molecular weight. Lane 1: protein ladder, lane 2: sea anemone venom. Please click here to view a larger version of this figure.
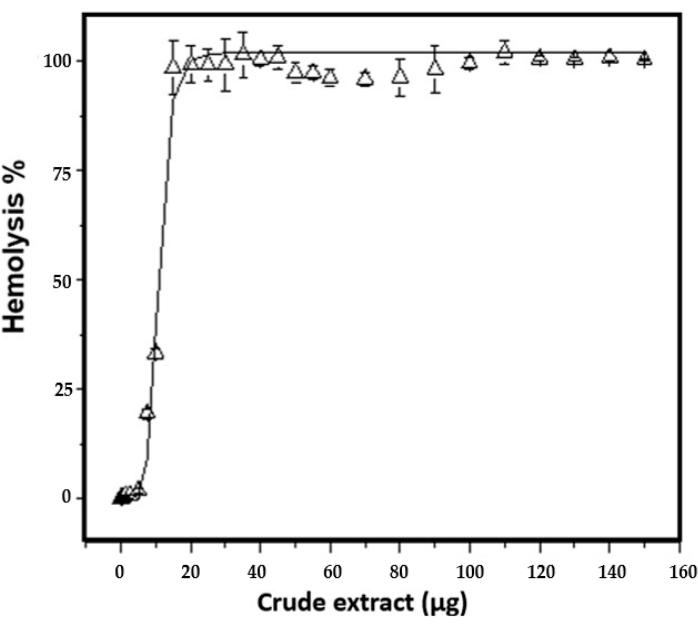
Figure 5: Hemolysis assay in erythrocytes from sheep. Hemoglobin release was detected at 415 nm. Different amount of protein was assayed to determine HU50, equal to 11.1 ± 0.3 µg/mL. Hemolysis assays were performed in triplicate. The bars represent the standard deviation. Please click here to view a larger version of this figure.
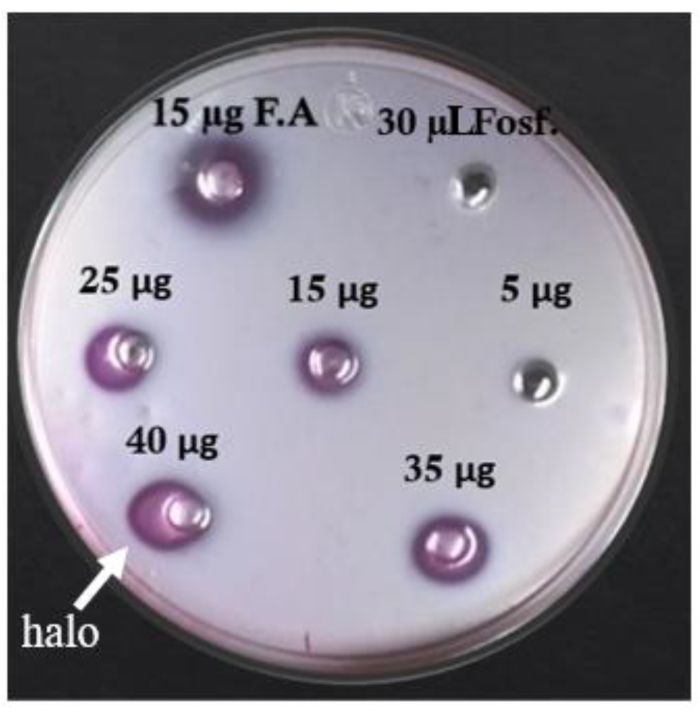
Figure 6: Phospholipase assay. Different amounts of total protein were assayed in agarose with egg yolk in the presence of Ca2+. Phospholipase activity can be observed around each well like a halo. Controls: phosphate buffer (Fosf.) and a PLA2 from a snake (F.A). Please click here to view a larger version of this figure.
| Tube | Distilled Water | BSA | Dye (µL) | Final volume | |
| (µL) | (µg) | (µL) | (µL) | ||
| 1 | 800 | – | – | 200 | 1000 |
| 2 | 799 | 1 | 1 | 200 | 1000 |
| 3 | 797 | 3 | 3 | 200 | 1000 |
| 4 | 795 | 5 | 5 | 200 | 1000 |
| 5 | 793 | 7 | 7 | 200 | 1000 |
| 6 | 790 | 10 | 10 | 200 | 1000 |
| Crude extract (µL) | |||||
| 7 | 798-790 | from 2 to 10 | 200 | 1000 | |
Table 1: Quantitation of protein by Bradford assay.
| Solution | Resolving gel | Stacking gel |
| Destilled water | 1.1 mL | 1.4 mL |
| Acrylamide mix (30%) | 2.5 mL | 0.33 mL |
| Tris (1.5 M, pH 8.8), adjust pH with HCl. | 1.3 mL | |
| Tris (0.5 M, pH 6.8), adjust pH with HCl. | 0.25 mL | |
| Sodium dodecyl sulfate (SDS) 10% (w/v) | 0.5 mL | 0.1 mL |
| Ammonium persulfate (APS) (10%) | 0.5 mL | 0.1 mL |
| N,N,Nˈ,Nˈ-tetramethylethylenediamine (TEMED) | 0.005 mL | 0.005 mL |
Table 2: Solutions for preparing a 15% acrylamide SDS-PAGE electrophoresis gel.
| Well | Alserver solution (µL) | Distilled water | Crude extract | Erythrocyte solution (µL) | Final volume | Expected absorbance |
| (µL) | (µL) | (µL) | (415 nm) | |||
| 1 | 180 | – | – | 20 | 200 | ≤0.1 |
| 2 | – | 180 | – | 20 | 200 | ≤1.0 |
| 3 | 177–170 | – | 3–10 | 20 | 200 | 0.1–1.0 |
Table 3: Hemolysis assay to calibrate erythrocytes.
| Sample | Diameter of halo (mm) |
| 15 µg of F.A | 0 |
| 30 µL of Fosf. | 6.3 ± 0.5 |
| 5 µg | 0 |
| 15 µg | 2.3 ± 0.5 |
| 25 µg | 3.5 ± 0.5 |
| 35 µg | 4.1 ± 1 |
| 45 µg | 4.6 ± 0.8 |
Table 4: Diameter of the halos at different amounts of protein from the crude extract.
