The results show plate images of raw and fluorescence images from ambient and low CO2 screening of WT and test mutants. Each plantlet is labeled by area number, with corresponding fluorescence readings given as QY. The data are exported as a text file and can be opened in a spreadsheet for analysis (see Supplemental Table S1). Mutant lines plgg1-1 and abcb26 were selected to demonstrate the positive and negative identification of genes associated with photorespiratory stress. PLGG1 codes for the first transporter in the pathway following the oxygenation of RuBP6, while ABCB26 is thought to code for an antigen transporter not known to be involved in photorespiration11. The dark-adapted Fv/Fm QY efficiencies of WT and mutants are visualized by box and whisker plots. To test the statistical difference between the WT and test mutants, a pairwise t-test was used with a p-value < 0.05. Here, we used photorespiratory mutant lines with reduced QY Fv/Fm as test mutants to check the efficiency of the screening method. The results show that the test mutants have a significantly lower QY efficiency than WT. Figure 3B shows a significant reduction in the ratio of variable to maximal fluorescence (Fv/Fm) for plgg1-1 but not abcb26 at low CO2. This result is consistent with the role of PLGG1 as a transporter involved in photorespiration, while abcb26 does not demonstrate a phenotype different from the WT control under low CO2 conditions. Thus, this screening method can identify photorespiratory mutants using low CO2 screening.
Figure 1: Example schematic of the seed plate layout. Two technical replicates are shown with six seeds placed in a row. WT control seeds placed above mutant seeds. Abbreviation: WT = wild type. Please click here to view a larger version of this figure.
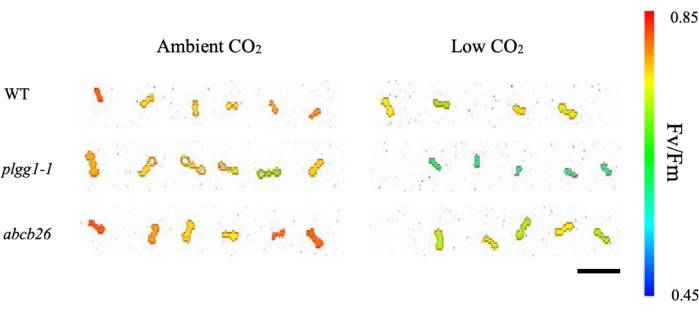
Figure 2: Dark-adapted Fv/Fm images of 9-day-old seedlings. The wild-type control is compared to T-DNA mutant lines at ambient and low CO2. The color scale represents the average Fv/Fm of each seedling. Scale bar = 1 cm. Abbreviations: Fv/Fm = ratio of variable to maximal fluorescence; WT = wild type; plgg1-1 = plastidal glycolate/glycerate translocator 1; abcb26 = ATP-binding cassette B26. Please click here to view a larger version of this figure.
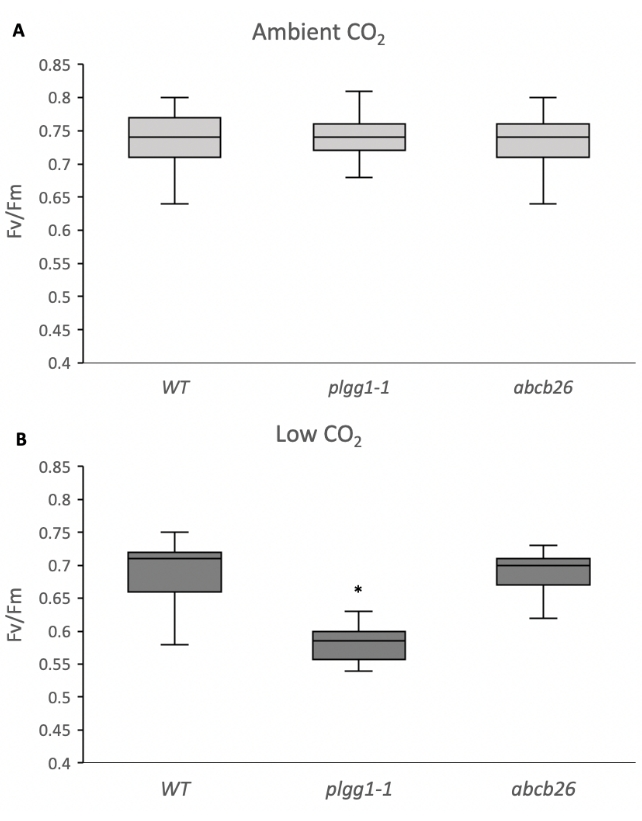
Figure 3: Fluorescence observations. Maximal quantum yield (Fv/Fm) measurements made on seedlings at (A) ambient and (B) low CO2 conditions. The boxes represent the range between the inner quartiles, the lines within the boxes represent the medians, and the whiskers represent the maximal and minimal observations. * Indicates significant difference based on a pairwise t-test relative to WT (n > 44, p < 0.05; Supplemental Table S2). Abbreviations: Fv/Fm = ratio of variable to maximal fluorescence; WT = wild type; plgg1-1 = plastidal glycolate/glycerate translocator 1; abcb26 = ATP-binding cassette B26. Please click here to view a larger version of this figure.
Supplemental Table S1: Compiled fluorescent data from all seedling plates used in the experiment. Please click here to download this File.
Supplemental Table S2: Statistics are reported from a t-test between wild type and both mutant genotypes. Mutants are considered significantly different from wild type when the p-value is lower that 0.05. Table A represents t-tests performed on plants grown in ambient CO2, while Table B represents t-tests performed on plants grown in low CO2. t-Test: two-sample assuming equal variances. Please click here to download this File.
