Clinical profile of the families
The family pedigrees of HCM were obtained and are shown in Figure 2. All the documented family members were diagnosed with HCM at enrollment.
In the family (Figure 2A), the proband was patient III-7, who was diagnosed with HCM and left ventricular outflow tract obstruction (LVOTO) at 46 years old and underwent cardiac surgery. Patient III-3 had minor HCM that did not require surgical treatment. Patient IV-3 also had minor HCM, which was similar to his father, Patient III-3. Patient II-5 had HCM and underwent surgery to repair the defect at the age of 51 due to LVOTO. Patient I-1 and patient II-2 died due to cardiac accidents at 57 and 46 years old, respectively. Patient I-1's medical history was unavailable. Patient II-7 and patient III-9 presented with a shortness of breath and were diagnosed with HCM.
Exome sequence analysis and segregation of variants
In this family, exome sequencing of the five individuals generated a mean of a total of 19,978,731 pairs of sequenced reads with an average read length of 125 bp. In total, 98.72% of sequenced reads passed the quality assessment and were mapped to 98.66% of the human reference genome. Even after filtering, more than 42 variants (including single nucleotide substitutions and indels) were shared by these four patients. Of these, 27 were missense SNVs, 15 were predicted to alter splicing. Finally, according to ACMG rating guidelines, a heterozygous c.G2468A; p.G823E variant of MYH7 (NM_0002571) was observed in proband III-7 as well as the other three patients.
Identification of a pathogenic mutation
Sanger sequencing confirmed the same MYH7 p.G823E variant in all patients but not in the healthy individuals in the families and 174 controls (Figure 2B). The heterozygous MYH7 p.G823E completely co-segregated in this family. In this family, patients IV-3 who harbored this variant inherited it from patient III-3. Patients III-7 and III-8 carried the same variant, which was inherited from their father. The information for patient III-9 was unavailable.
This variant, previously described in HCM by a sporadic patient24, has not been reported in the 1000 G, ESP6500, ExAC, HGMD, ClinVar databases or control subjects. The glycine residue at codon 823 in the neck domain region of MYH7 is highly conserved in all the available vertebrate myosin sequences (Figure 2C). MYH7 is a known HCM-causative gene and plays an important role in cardiac development or structure/function. Based on ACMG standards and guidelines, the MYH7 p.G823E variant was predicted to be a pathogenic variant (PVS1 + PS3 + PS4 + PM2). Taken together, these results support that this variant is detrimental and contributes to the pathogenesis of HCM in these families.
C57BL/6N-Myh7em1(G823E) knockin mice developed severe cardiac hypertrophy
To further verify the pathogenesis of MYH7 p.G823E, we generate C57BL/6N-Myh7em1(G823E) knockin mice. C57BL/6N-MYH7em1(G823E) knockin mice developed an age-dependent cardiac hypertrophy after birth (Figure 2A,B). Echocardiography revealed that IVS and LVPW developed with increased HR in C57BL/6N-Myh7em1(G823E) knockin mice (Table 1). These results were likewise validated by histological analysis (Figure 3). There were no obvious differences in LVDd, LVDs, EF, or CO between wild-type and heterozygous mice (Table 1).
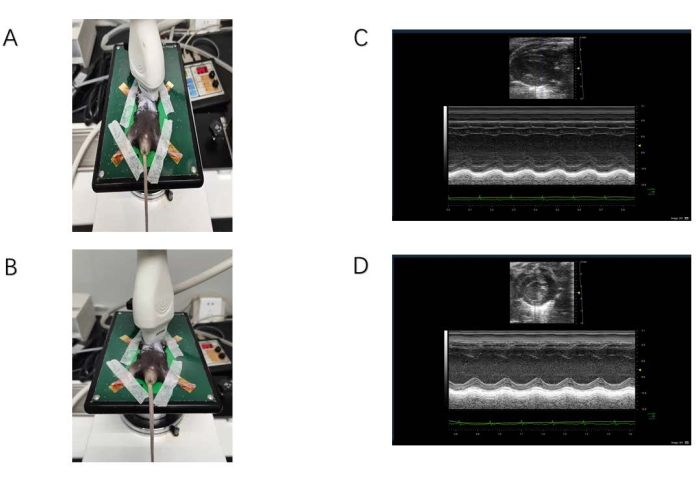
Figure 1: Ultrasound data acquisition. (A) The probe is located on the long axis of the sternum. (B) The probe is located on the short axis of the sternum. (C) M-mode ultrasound image of the long-axis view of the sternum. The yellow arrow indicates the location of the interventricular septum. The red arrow indicates the floating valve. (D) M-mode ultrasound image of the short-axis view of the sternum. The yellow arrow indicates the location of the anterior papillary muscle, and the bulge below is the posterior papillary muscle. Please click here to view a larger version of this figure.
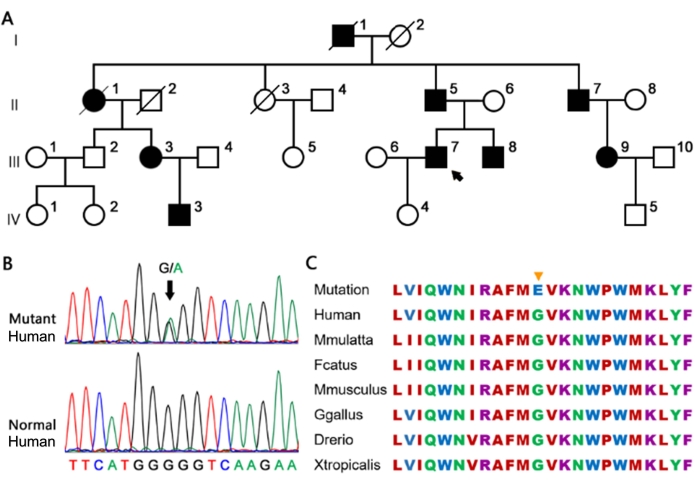
Figure 2: The large family carrying the heterozygous MYH7 G823E variant. (A) The proband is marked by an arrow. Full and open circles and squares indicate affected and normal individuals, respectively. (B) The G823E variant in MYH7 confirmed by Sanger sequencing. (C) Conservation of the MYH7 G823E site in different species. The yellow arrow represents the site of G823E in the amino acid sequence of different species, which is highly conserved. Please click here to view a larger version of this figure.
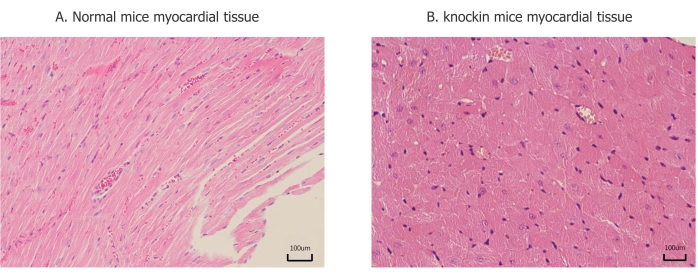
Figure 3: Pathological changes of myocardial tissue. (A) Myocardial fibers are arranged in an orderly manner, and there is no significant difference between each part. (B) Ventricular muscle fiber hypertrophy, disordered arrangement, and lesions are mainly concentrated in the posterior wall of the left ventricle and interventricular septum. Please click here to view a larger version of this figure.
| C57BL/6N-Myh7em1(G823E) knockin mice group | Control group | ||
| (n=4) | (n=6) | ||
| HR (/min) | 451.25 ± 25.786 | 413.83 ± 12.77 | P = 0.015 |
| LVDd (mm) | 4.12 ± 0.33 | 3.95 ± 0.20 | P = 0.330 |
| LVDs (mm) | 2.95 ± 0.44 | 2.85 ± 0.20 | P = 0.626 |
| EF(%) | 55.02 ± 9.52 | 54.31 ± 5.11 | P = 0.881 |
| CO (mL) | 18.46 ± 3.05 | 15.30 ± 2.39 | P = 0.102 |
| IVS (mm) | 1.13 ± 0.20 | 0.67 ± 0.07 | P = 0.001 |
| LVPW (mm) | 1.40 ± 0.60 | 0.70 ± 0.06 | P = 0.000 |
Table 1: Morphology and function analysis for p.G823E and wild type. Data were analyzed using SPSS. Continuous variables are expressed as mean ± standard deviation (SD). The Student's t or Wilcoxon rank sum tests for continuous variables. P-values below 0.05 were considered statistically significant.
Supplemental File 1. Please click here to download this File.
