Representative recordings obtained using the method described here are presented. In vivo Ca2+ imaging using holographic microscopy requires 2-4 weeks to complete from head plate implantation and AAV injection to data acquisition. Therefore, to obtain stable results, it is important to reduce motion artifacts in the brain. The head plate implantation (step 1.5) and the placement of the cranial window (step 2.9) are very important steps in this process. Furthermore, it is also important to select an AAV (AAV2/8-CaMKII-GCaMP6m-P2A-ChRmine-Kv2.1) that simultaneously expresses calcium indicator and opsin in a single neuron (step 2.8).
Figure 4A shows a spot for the holographic illumination of an acquired neuron image using a custom-made MATLAB script. If holographic illumination successfully illuminated the neurons expressing jGCaMP8f, Ca2+ traces could be obtained with an image sensor (Figure 4B), as shown in Figure 4C.
Functional connectivity between neurons was evaluated using holographic stimulation (Figure 4D), as shown in Figure 4E. Because functional connectivity between neurons is one of the neural circuitry properties that is altered in the pathogenesis of a pain model mouse24, we describe a simple procedure to evaluate it. Figure 4F shows a typical image of L2/3 neurons in S1HL visualized using GCaMP6m. When one neuron (orange circle) was holographically stimulated, another neuron (red circle) was simultaneously active; thus, the number of functional connectivities between neurons was one (Figure 4G).
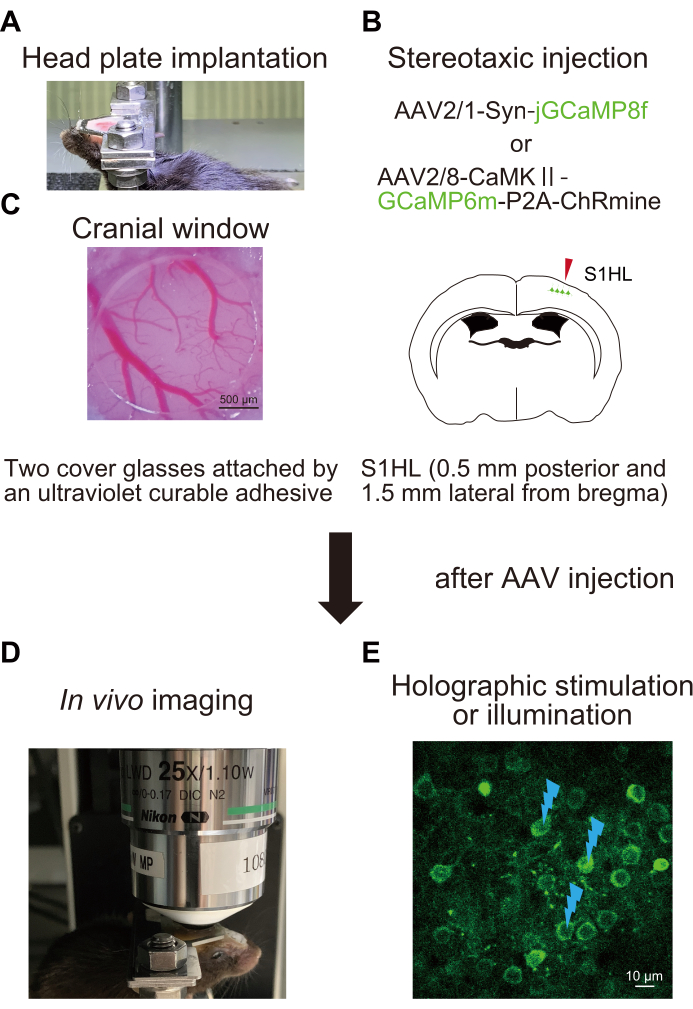
Figure 1: Schematic outline of the experimental procedure. (A) Fixation of the head plate to the skull. (B) Stereotactic injection of AAV into the hind paw area of the primary somatosensory cortex (S1HL). (C) Implantation of the cranial window. To assess and manipulate neural activity, in vivo Ca2+ imaging is performed in awake mice (D) with holographic stimulation (E). Flash marks indicate holographic stimulation or illumination. Please click here to view a larger version of this figure.
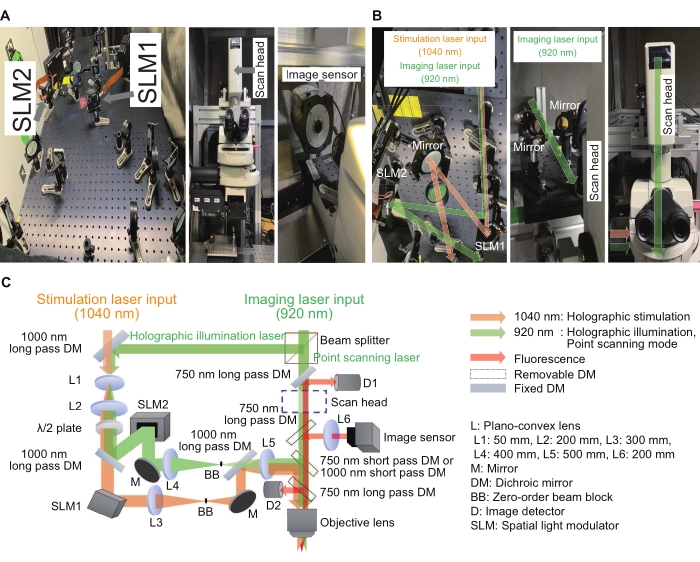
Figure 2: System used for the holographic microscope. (A) Images of the holographic stimulation and illumination light paths (left) near the microscope (middle) with an image sensor (right). (B) These are magnified images of holographic stimulation and illumination light paths around respective SLMs (left and right) and a point scanning light path around a scan head (middle and right). (C) A schematic of the stimulation and imaging optical paths. Phase-only SLMs are used to display digital holograms, and a beam expander (a combination of L1 and L2) and a 4f relay system (a combination of L3 and L4 for holographic stimulation and L4 and L5 for holographic illumination) are placed before and after respective SLMs to make sure each digital hologram is imaged at the exit pupil of a water immersion objective lens, with slightly under-filled image size. To suppress residual zero-order components, a beam block is placed at the intermediate plane. Please click here to view a larger version of this figure.
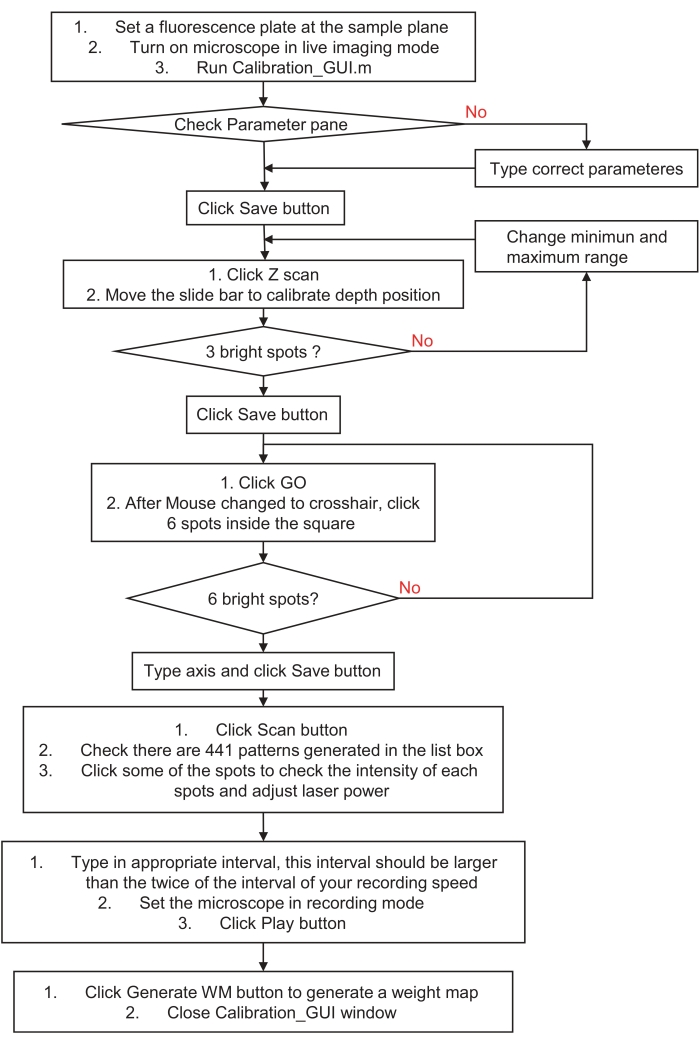
Figure 3: Flowchart of how to calibrate the holographic stimulation or illumination system. This flowchart describes steps to calibrate a holographic stimulation or illumination system to the sample space and imaging system. Please visit step 3, preparation for the holographic stimulation or illumination system, for detailed instructions and download a sample program. Please click here to view a larger version of this figure.
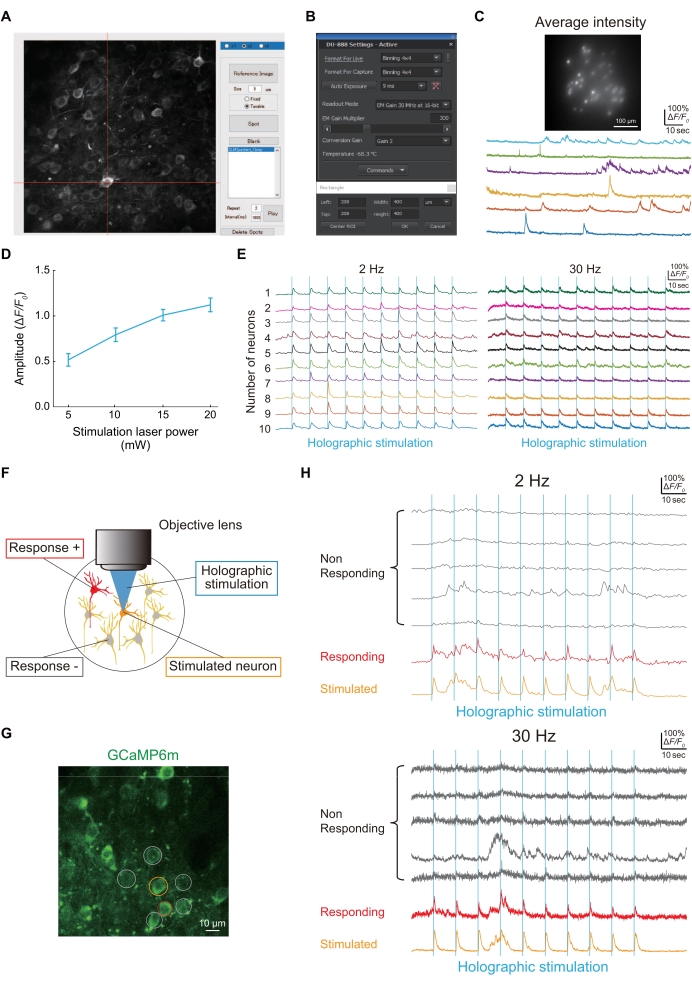
Figure 4: Representative results of imaging and functional connectivity using a holographic microscope. (A) To illuminate specific neurons expressing jGCaMP8f or GCaMP6m-P2A-ChRmine, the image of neurons is captured, and then a spot is formed on the neuron using a custom-made MATLAB script. (B) The setting of the image sensor (exposure time, imaging area, and binning). (C) Representative image and traces of neurons expressing jGCaMP8f in 100 Hz imaging with holographic illumination and an image sensor. (D) This graph shows the neural response to holographic stimulation (1,040 nm) at each laser power (data from GCaMP6m-P2A-ChRmine expressing neurons at 2 Hz imaging frame rate [n = 16]). Error bars indicate the standard error of the mean. (E) Representative Ca2+ traces during holographic stimulation (blue vertical lines) of 10 different neurons at 2 Hz (left) and 30 Hz (right) imaging frame rates. In 2 Hz and 30 Hz Ca2+ traces, the same color indicates the same neuron. (F) Schematic diagram evaluating functional connections between neurons. When the orange neuron is stimulated, the red neurons respond at the same time, indicating that there is functional connectivity between these neurons. (G) A typical image of S1HL neurons expressing GCaMP6m in WT. Scale bar = 10 µm. (H) Typical Ca2+ traces during holographic stimulation (blue vertical lines) at 2 Hz (upper) and 30 Hz (lower) imaging frame rates. The stimulated neuron is circled in orange, responding neurons are circled in red, and non-responding neurons are circled in grey. Neural responsiveness to holographic stimulation can be detected at both 2 Hz and 30 Hz imaging speeds. Please click here to view a larger version of this figure.
| Our set-up 6 | Prakash, R. et al. 25 | Marshel, J. H. et al. 12 | Robinson, N. T. M. et al. 14 | |
| Lateral resolution | 1.2 μm | 1.27 μm | ― | 2.22 μm |
| Axial resolution | 8.3 μm | 56.86 μm | 15.5 μm | 10.26 μm |
| Objective lens/Numerical Aperture | 25x/1.1 | 20x/0.5 | 16x/0.8 | 16x/0.8 |
Table 1: Summary of previous reports and this system for the spatial resolution of holographic stimulation. The lateral resolution, axial resolution, and the objective lens used during the measurement are described.
