Visualizing DNA Damage Repair Proteins in Patient-Derived Ovarian Cancer Organoids via Immunofluorescence Assays
Summary
The present protocol describes methods for evaluating DNA damage repair proteins in patient-derived ovarian cancer organoids. Included here are comprehensive plating and staining methods, as well as detailed, objective quantification procedures.
Abstract
Immunofluorescence is one of the most widely used techniques to visualize target antigens with high sensitivity and specificity, allowing for the accurate identification and localization of proteins, glycans, and small molecules. While this technique is well-established in two-dimensional (2D) cell culture, less is known about its use in three-dimensional (3D) cell models. Ovarian cancer organoids are 3D tumor models that recapitulate tumor cell clonal heterogeneity, the tumor microenvironment, and cell-cell and cell-matrix interactions. Thus, they are superior to cell lines for the evaluation of drug sensitivity and functional biomarkers. Therefore, the ability to utilize immunofluorescence on primary ovarian cancer organoids is extremely beneficial in understanding the biology of this cancer. The current study describes the technique of immunofluorescence to detect DNA damage repair proteins in high-grade serous patient-derived ovarian cancer organoids (PDOs). After exposing the PDOs to ionizing radiation, immunofluorescence is performed on intact organoids to evaluate nuclear proteins as foci. Images are collected using z-stack imaging on confocal microscopy and analyzed using automated foci counting software. The described methods allow for the analysis of temporal and special recruitment of DNA damage repair proteins and colocalization of these proteins with cell-cycle markers.
Introduction
Ovarian cancer is the leading cause of death due to gynecologic malignancy. The majority of patients are treated with DNA-damaging drugs such as carboplatin, and those with homologous recombination repair (HRR)-deficient tumors can be given poly (ADP-ribose) polymerase (PARP) inhibitors1,2. However, most patients develop resistance to these therapies and die within 5 years of diagnosis. Dysregulation of the DNA damage response (DDR) has been associated with the development of ovarian cancer and both chemotherapy and PARP inhibitor resistance3. Thus, study of the DDR is imperative in understanding the pathophysiology of ovarian cancer, potential biomarkers, and novel targeted therapies.
Current methods to evaluate the DDR utilize immunofluorescence (IF), as this allows for the accurate identification and localization of DNA damage proteins and nucleotide analogs. Once there is a double-stranded break (DSB) in the DNA, the histone protein H2AX is rapidly phosphorylated, forming a focus where DNA damage repair proteins congregate4. This phosphorylation can be easily identified utilizing IF; in fact, the ɣ-H2AX assay has commonly been employed to confirm the induction of a DSB5,6,7,8,9. Increased DNA damage has been associated with platinum sensitivity and efficacy of DNA damaging agents10,11,12, and ɣ-H2AX has been proposed as a biomarker associated with chemotherapy response in other cancer treatments13. Upon a DSB, a cell proficient in HRR performs a series of events that leads to BRCA1 and BRCA2 recruiting RAD51 to replace replication protein A (RPA) and bind to the DNA. HRR repair uses a DNA template to faithfully repair the DSB. However, when tumors are deficient in HRR, they rely on alternative repair pathways such as non-homologous end joining (NHEJ). NHEJ is known to be error-prone and creates a high mutational burden on the cell, which uses 53BP1 as a positive regulator14. These DNA damage proteins can all be accurately identified as foci using IF. In addition to staining for proteins, IF can be used to study fork protection and single-stranded DNA gap formation. The ability to have stable forks has been correlated with platinum response, and recently, gap assays have shown the potential to predict the response to PARP inhibitors6,15,16,17. Therefore, staining for the nucleotide analogs after introduction into the genome is another way to study the DDR.
To date, evaluation of the DDR in ovarian cancer has been largely limited to homogenous 2D cell lines that do not recapitulate the clonal heterogeneity, microenvironment, or architecture of in vivo tumors18,19. Recent research suggests organoids are superior to 2D cell lines in studying complex biologic processes such as DDR mechanisms6. The present methodology evaluates RAD51, ɣ-H2AX, 53BP1, RPA, and geminin in PDOs. These methods assess the intact organoid and allow for the study of DDR mechanisms in a setting more similar to the in vivo tumor microenvironment. Together with confocal microscopy and automated foci counting, this methodology can aid in understanding the DDR pathway in ovarian cancer and personalizing treatment plans for patients.
Protocol
Tumor tissue and ascites were obtained after obtaining patient consent as part of a gynecologic oncology biorepository that was approved by the Washington University in St. Louis Institutional Review Board (IRB). Patients were included if they had advanced stage high-grade serous ovarian cancer (HGSOC). All procedures were performed at room temperature on the bench unless otherwise specified. All reagents were prepared at room temperature (unless otherwise indicated) and stored at 4 °C.
1. Organoid generation
- Generate the organoids following the previously published report20.
2. Plating and irradiation of organoids
- Using organoids in culture, dislodge the tabs of the organoids from the culture dish through manual manipulation of a pipette tip. Collect the organoids in a 15 mL conical tube.
- Centrifuge the conical tube at 1,650 x g for 5 min at 4 °C. Using a pipette, aspirate off the supernatant carefully.
- Add 1,000 µL of an animal origin-free, recombinant enzyme to the pellet (see Table of Materials). Mix the solution and incubate for 15 min at 37 °C.
- Centrifuge the solution at 1,650 x g for 5 min at 4 °C. Using a pipette, carefully aspirate off the supernatant.
- After centrifugation and aspiration, resuspend the pellet in 1,000 µL of phosphate-buffered saline.
- Transfer 10 µL of the solution to a 1.5 mL microcentrifuge tube and mix with 10 µL of trypan blue.
- Take 10 µL of the trypan blue cell solution into a cell counting chamber slide and insert it into the cell counting machine (see Table of Materials).
- Plate 40,000 cells per 20 µL of basement membrane extract (BME) into an 8-well glass chamber slide (see Table of Materials). This maintains the organoids for 3-5 days to allow the cells to form organoids and grow to a suitable size.
- To induce double-strand DNA breaks, radiate the plated organoids with 10 Gray (Gy) of ɣ-irradiation (see Table of Materials for irradiator details).
NOTE: This could take up to 5-10 min. - Incubate the organoids in their culture media in a humidified incubator at 37 °C and 5% CO2 for 4 h.
3. Immunofluorescence staining
NOTE: Volumes refer to the amount per well of the 8-well chamber slide (~300 µL).
- After irradiation and incubation of the organoids, aspirate the media with a pipette, carefully so as not to disrupt the 3D matrix. Wash with 300 µL of PBS.
- Fix the organoids in 300 µL of 2% paraformaldehyde (PFA) for 10 min.
NOTES: Do not leave for too long, as the BME will depolymerize and could be aspirated off. - Wash the organoids with 300 µL of staining buffer (see Table of Materials). Place on a shaker for 5 min.
- For permeabilization, gently add 300 µL of permeabilization buffer (see Table of Materials) and incubate for 20 min. Wash with 300 µL of staining buffer. Place on a shaker for 5 min.
- Add 300 µL of staining buffer to block the permeabilization step. Place on a shaker for 30 min. Aspirate off the staining buffer using a pipette.
- Add 300 µL of primary antibodies (rabbit anti-RAD51, mouse anti-Geminin, rabbit anti-RPA, mouse anti-ɣH2AX, rabbit anti-Geminin, mouse anti-53BP1; see Table of Materials) diluted in staining buffer. Incubate at 4 °C for 16 h.
NOTE: Avoid co-staining with the same host animal. Leave the lid off to avoid the antibodies mixing into neighboring wells. - Remove the primary antibody solution. Perform three washes with 300 µL of staining buffer for 5 min each on the shaker.
- Add 300 µL of secondary antibody (goat anti-mouse Alexa fluor 488 and goat anti-rabbit Alexa fluor 647; see Table of Materials) solution diluted in staining buffer and incubate for 1 h in the dark.
NOTE: All subsequent steps must be performed in the dark. - Aspirate the secondary antibody solution. Add 300 µL of diluted DAPI. Place on a shaker for 5 min.
- Perform three washes with 300 µL of staining buffer for 5 min each on the shaker. Remove the staining buffer and detach the chambers using the removal kit included with the chamber slides (see Table of Materials).
NOTE: Ensure not to push too far forward so as to disrupt the 3D matrix. - Using a p200 pipette tip with the tip cut off, add mounting medium (see Table of Materials) to cover each well with an organoid tab (e.g., 20-30 µL for each organoid tab).
- Place a coverslip over the specimen, avoiding bubbles.
- Take clear nail polish and paint it onto the sides of the cover glass to seal the slide. Let it harden for 1 h and place it at -20 °C.
NOTE: After imaging, the slide can be stored for at least 6 months with minimal loss of fluorescence.
4. Imaging
- Acquire images using a confocal microscope (see Table of Materials) at 63x objective with immersion oil.
NOTE: If imaging nuclear proteins, 63x is advantageous, but 40x is also acceptable. - Use DAPI to locate the organoids through the eyepiece. Select the appropriate filters for microscopy based on the fluorophores used. Set the parameters for capturing the z-stack images.
NOTE: The fluorophores used in the study were DAPI, 488, and 647. The 405, 488, and 638 lasers were turned on to visualize the staining.
NOTE: The recommended z-stack depends on the resolving power of the objective. - Adjust the live image: set focus, laser intensity, etc.
NOTE: The higher the laser intensity, the more likely the sample will photo-bleach. Therefore, select an optimum laser intensity. - Acquire the z-stack.
NOTE: This could take up to 5-10 min. - From the images collected in the z-stack, screenshot at least three images per stack.
NOTE: Ensure to obtain screenshots throughout the stack so as to not duplicate the nuclei when quantifying. - Save each file as a TIFF and proceed to analysis (step 5).
5. Analysis
NOTE: Use JCountPro for all image analysis following the previously published report21. To obtain this software, reference the associated publication.
- Under the object analysis panel (top of the program) in the file selection tab (bottom of the program), select the TIFF files.
- Click Add to move the selected files to the selected files group. Select Display.
- Under the segmentation tab, select blue as the color of the nuclei. Select Auto segmentation to optimize the threshold of the nuclei.
NOTE: If the auto segmentation does not accurately identify the objects, then the user can adjust the fine tune and raw tune to the image or see the user manual to further optimize the segmentation. - Under the objects panel, select Auto split large objects and optimize the size of the nuclei.
NOTE: The nuclei typically have an unconditional size of 5,000 pixels, while the conditional size is 1,500 pixels. See the user manual to further optimize the size of the objects. - Select Identify objects to test the parameters.
NOTE: If these parameters are not accurate, the size and tuning can be adjusted along with other settings. See the user manual for instructions regarding further optimization. - After adjusting the parameters to the image, select Identify Objects in All Images to identify the nuclei in all the images selected.
- Select the Foci Analysis panel (top of the program).
- Under the select files tab (bottom of the program), select the TIFF files that were used for object analysis and select the Arrow Buttons (>>) to move the images to the image files group.
- In the directory group underneath the image files that were moved, double click the Manual Edit folder and select to move the ioc files into the object collection files group.
NOTE: All TIFFs selected must have matched ioc files. - Press Select to move the files into the selected files group. Select the Foci counting tab at the bottom of the program.
- Optimize the parameters following the steps below.
- Top hat settings index: 12.
- Focus channel: Select the color of the foci (green: ɣ-H2AX; red: RAD51).
- H dome settings: dome height %: 30; threshold %: 28 and 28.
- Shape/size setting: Min focus size, pixels: 5. Select Apply Shape/Size. Max focus size (conditional): 32. Min roundness, x100: 96. Max focus, pixels: 60.
- Noise filter: size 1.
NOTE: If determining if a nucleus is positive for geminin staining, under the second channel select green, and under analysis select intensity.
- To test the parameters set, select the below buttons in order: Top Hat > H-Dome > Foci Count.
NOTE: If these parameters do not work, the size and tuning can be adjusted along with other settings. See user manual to learn more about how the image can be further optimized. - Once the parameters are optimized for the selected images, select Auto count to count the foci in each image per nuclei. If the second channel is desired, the program will determine the intensity which can be used.
Representative Results
The presented protocol can successfully stain, visualize, and quantify nuclear DNA damage repair proteins in organoids. This technique was used to stain and evaluate PDOs both before and after irradiation. PDOs were exposed to 10 Gy of radiation and evaluated for the following biomarkers: ɣ-H2AX (Figure 1), a marker of DNA damage; RAD51 (Figure 2), a marker for HRR; 53BP1, a marker of NHEJ; RPA, a marker of replication stress (Figure 3); and geminin, a G2/S phase cell cycle marker14. The dose of 10 Gy was chosen based on previously published research studying DNA damage in ovarian cancer6,22. JCountPro software was used to identify the nucleus and quantify the number of foci within the nucleus with illustrated parameters13 (Figure 4). The software identifies the nucleus (Figure 4A,B), then nuclear foci (Figure 4C), and filters the foci from geminin-positive cells (Figure 4D).
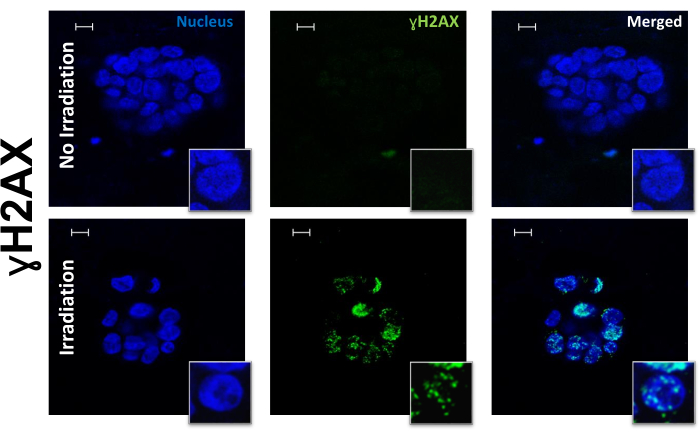
Figure 1: ɣ-H2AX foci in PDOs before and after irradiation. Representative images of DAPI and ɣ-H2AX foci in PDOs before and after irradiation at 10x with 63x insets. Scale bars: 10 µm. Please click here to view a larger version of this figure.
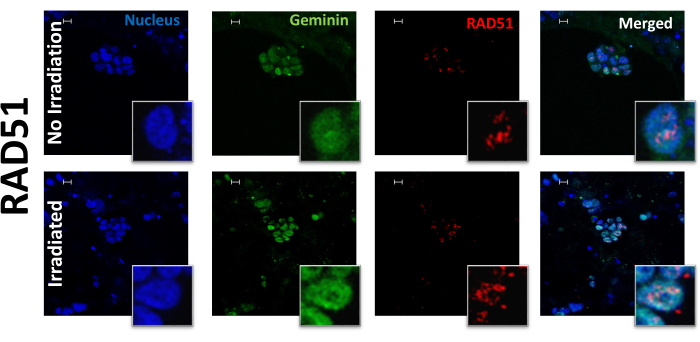
Figure 2: RAD51 foci and geminin in PDOs before and after irradiation. Representative images of DAPI, geminin, RAD51, and co-staining of geminin/RAD51 foci PDOs before and after irradiation at 10x with 63x insets. Scale bars: 10 µm. Please click here to view a larger version of this figure.
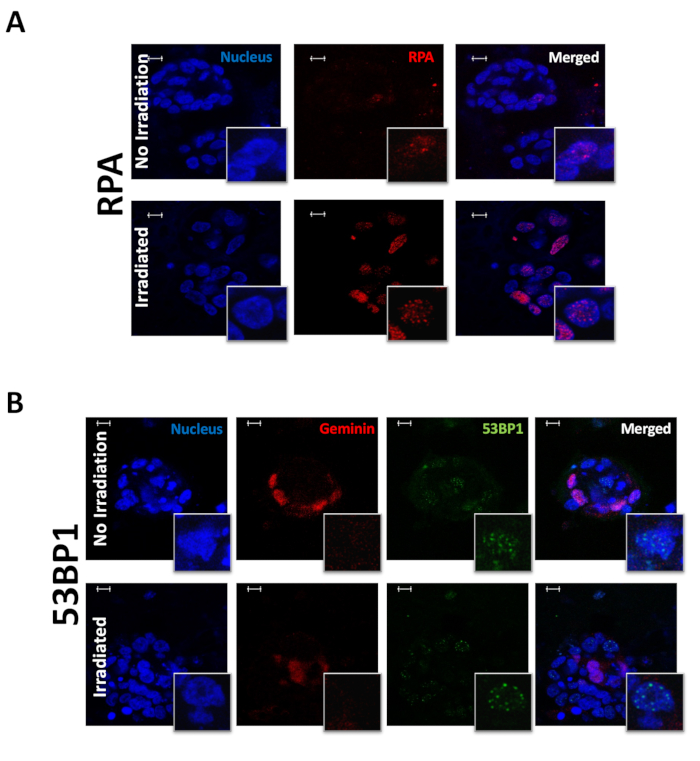
Figure 3: RPA and 53BP1 in PDOs before and after irradiation. Representative images of (A) DAPI, RPA; (B) geminin, 53BP1, and co-staining of geminin/53BP1 foci at 10x with 63x insets. Scale bars: 10 µm. Please click here to view a larger version of this figure.
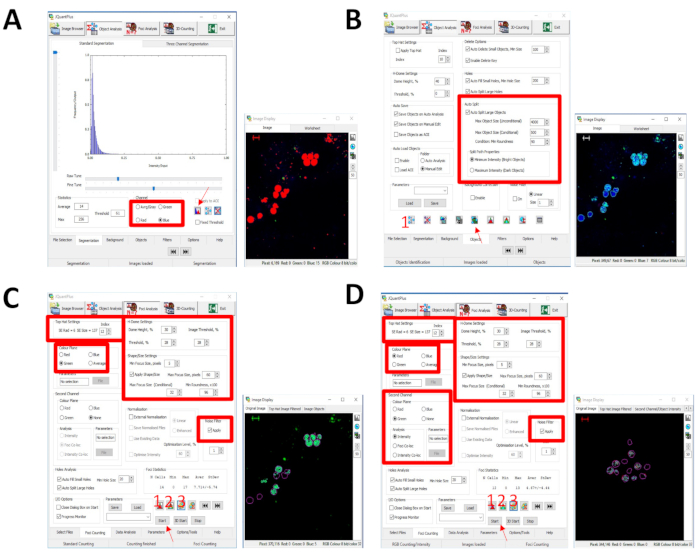
Figure 4: The JCountPro software quantification workflow. (A) Under the object analysis, the blue channel is selected to identify the blue objects, nuclei, then it is automatically optimized by selecting the auto segmentation (red arrow). (B) Under auto split, the object size (nuclei) is adapted to the size of the image and magnification. The identify object button is selected to test the parameters (1), and the identify objects in all images (red arrow) button is selected to identify objects (nuclei) for each image. (C) Under the foci analysis tab, the images are inputted and the foci counting parameters are set: first, the color of the foci is selected under the focus channel, green; the top hat index is set to 12; the H dome settings are set to have a dome height percentage of 30 and a threshold percentage of 28; the shape and size of the foci are optimized to the image size with the manual parameters of maximum focus size pixels at 60 and minimum roundness x 100 at 96; finally, the noise filter is applied. The settings are tested by applying the top hat, H dome, and foci count (1-3). To quantify the ɣ-H2AX foci per cell, press start (red arrow). (D) For the RAD51 foci, the settings are applied as illustrated for ɣ-H2AX foci, except the focus channel which is changed to red; however, to identify nuclei that are stained with geminin, the second channel is selected for green, and analysis is changed to intensity. The parameters are tested by applying the top hat, H dome, and foci count (1-3), then pressing start (red arrow) to quantify all RAD51 foci and evaluating the intensity of green per cell in each image. Scale bar: 10 µm. Please click here to view a larger version of this figure.
Discussion
The DNA damage response plays an integral role in both the development of ovarian cancer and chemotherapy resistance. Therefore, a thorough understanding of DNA repair mechanisms is imperative. Here, a methodology is presented to study DNA damage repair proteins in 3D, intact organoids. A reproducible, reliable protocol is developed utilizing hallmark antibodies to evaluate DNA damage, homologous recombination, non-homologous end joining, and replication stress. Importantly, these methods are validated using genetically modified controls demonstrating the specificity and sensitivity of these methods.
The most critical step in this protocol is the plating and fixation of the organoids. This protocol specifically incubates organoids in 2% PFA for 10 min with close monitoring of the basement membrane extract (BME) during the fixation process. This differs from other immunofluorescence protocols in organoids that suggest using 4% PFA for 10-60 min23,24,25. It has been found that if the tabs of the BME are in a concentrated amount of PFA for too long, the tabs depolymerize and are subject to aspiration. Of note, manufacturers of specific BMEs often offer suggestions regarding fixation. Another important point of discussion is the plating of the organoids. The staining process can result in losing a portion or all of the organoids. Therefore, the more confluent the tab at the time of plating, the higher likelihood the sample will successfully withstand the steps of staining.
The strengths of this protocol include the validation of nuclear antibody staining, the ability to perform immunofluorescence and imaging of intact 3D organoids, and the adaptability of the protocol to any treatment or molecule of interest.
The antibodies used in these experiments were stringently validated for specificity using genetically modified ovarian cancer cells. The methods were further validated for sensitivity by exposing PDOs to increasing doses of radiation. This resulted in increasing ɣ-H2AX, RAD51, and RPA foci in HRR-proficient PDOs. Lastly, the objective quantification methods facilitate the reproducibility of this assay and avoid the subjective bias of manual quantification.
Traditional methods to explore the DDR are through 2D culturing conditions known as cell lines, but they do not have the ability to maintain the genetic heterogeneity of the original tumor. The tumor microenvironment is vital in tumorigenesis and chemotherapy resistance in ovarian cancer26,27,28, therefore, these models offer an advantage to 2D homogenous cell cultures when studying the DDR. When studying the DDR, it is necessary to control for cell cycle in order to avoid misclassification of the inability to perform specific types of DNA repair that are cell cycle specific (i.e., HRR). Future work is focused on studying specific types of DNA lesions caused by platinum chemotherapy, poly (ADP-ribose) polymerase inhibitors, or hydroxyurea.
Lastly, this protocol is adaptable to study any antigen amendable to traditional immunofluorescence and can accommodate the study of novel drug therapies. The protocol presented focuses on DDR proteins, but with a simple change of antibodies, this protocol can be modified to study other proteins, glycans, and small molecules. Additionally, as the organoids are heterogenous 3D models cultured in vitro, any treatment of the organoids is possible, including immunotherapy, antiangiogenic therapy, metabolomics therapy, and other novel therapies that are difficult to evaluate in homogenous cell lines29,30,31.
Limitations of this method include using a BME with inconsistent composition and non-specific staining. As commercial BMEs are produced from cell lines, the composition of each unit can vary tremendously. These differences could affect the fixation and staining, as well as organoid generation. Additionally, depending on the sample, there can be non-specific staining of the BME, which can obstruct the protein of interest. Nonetheless, the nuclear staining is very specific and demonstrates clear nuclear foci which can be easily quantified.
In conclusion, a detailed protocol to evaluate DDR proteins in organoids is presented. As technology advances, it is anticipated that organoids will be able to be used to evaluate live cell DNA damage repair in these 3D models. Additionally, as the most effective method to culture, the organoids will be established, and more sophisticated assays such as DNA fiber and replication gap assays will be possible32.
Declarações
The authors have nothing to disclose.
Acknowledgements
We are grateful for the guidance of Pavel Lobachevsky, PhD in establishing this protocol. We'd also like to acknowledge Washington University's School of Medicine in St. Louis's Department of Obstetrics and Gynecology and Division of Gynecologic Oncology, Washington University's Dean's Scholar Program, Gynecologic Oncology Group Foundation, and the Reproductive Scientist Development Program for their support of this project.
Materials
| 1x phosphate buffered saline with calcium and magnesium (PBS++) | Sigma | 14-040-133 | |
| 1x phosphate buffered saline without calcium and magnesium (PBS) | Fisher Scientific | ICN1860454 | |
| Ant-53BP1 Antibody | BD Biosciences | 612522 | diluted to 1:500 in staining buffer |
| Ant-Geminin Antibody | Abcam | ab104306 | diluted to 1:200 in staining buffer |
| Anti-Geminin Antibody | ProteinTech | 10802-1-AP | diluted to 1:400 in staining buffer |
| Anti-RAD51 Antibody | Abcam | ab133534 | diluted to 1:1000 in staining buffer |
| Anti-yH2AX Antibody | Millipore-Sigma | 05-636 | diluted to 1:500 in staining buffer |
| Ant-phospho-RPA32 (S4/S8) Antibody | Bethyl Laboratories | A300-245A-M | diluted to 1:200 in staining buffer |
| Bovine Serum Albumin (BSA) | Fisher Scientific | BP1605 100 | |
| Centrifuge; Sorvall St 16R Centrifuge | Thermo Scientific | 75004240 | |
| Confocal Microscope, Leica SP5 confocal system DMI4000 | Leica | 389584 | |
| Conical Tubes, 15 mL | Corning | 14-959-53A | |
| Countess 3 FL Automated Cell Counter (Cell Counting Machine) | Thermo Scientific | AMQAF2000 | |
| Countess Cell Counting Chamber Slides | Thermo Scientific | C10228 | |
| Cover Slip | LA Colors | Any clear nail polish will suffice | |
| Cultrex RGF Basement Membrane Extract, Type 2 | R&D Systems | 3533-010-02 | Could probably use Matrigel or other BME Matrix |
| DAPI | Thermo Scientific | R37606 | NucBlue Fixed Cell ReadyProbes Reagent, Diluted in 1x PBS |
| Glycine | Fisher Scientific | NC0756056 | |
| JCountPro | JCountPro | For access to the software, Email: jcountpro@gmail.com | |
| Microcentrifuge Tubes | Fisher Scientific | 07-000-243 | |
| Nail Polish | StatLab | SL102450 | |
| Parafomraldehyde (PFA), 2% | Electron Microscopy Sciences | 157-4 | Dilute to 4% PFA in PBS++ to obtain 2% PFA |
| Permeabilization Buffer | Made in Lab | 0.2% X-100 Triton in PBS++ | |
| Pipette | Rainin | 17014382 | |
| Pipette Tips | Rainin | 17014967 | |
| ProLong Gold Antifade Mountant | Thermo Scientific | P36930 | |
| Staining Buffer | Made in Lab | 0.5% BSA, 0.15% Glycine, 0.1% X-100 Triton in PBS++ | |
| Thermo Scientific Nunc Lab-Tek II Chamber Slide System | Thermo Scientific | 12-565-8 | |
| Triton X-100 | Sigma-Alderich | 11332481001 | |
| Trypan Blue Solution, 0.4% | Thermo Scientific | 15250061 | |
| TrypLE Express | Invitrogen | 12604013 | animal origin-free, recombinant enzyme |
| X-RAD 320 Biological Irradiator | Precision X-Ray Irradiation | X-RAD320 |
Referências
- Lheureux, S., Gourley, C., Vergote, I., Oza, A. M. Epithelial ovarian cancer. Lancet Oncology. 393 (10177), 1240-1253 (2019).
- Tew, W. P., et al. PARP inhibitors in the management of ovarian cancer: ASCO guideline. Journal of Clinical Oncology. 38 (30), 3468-3493 (2020).
- Tomasova, K., et al. DNA repair and ovarian carcinogenesis: impact on risk, prognosis and therapy outcome. Cancers. 12 (7), 1713 (2020).
- Mukhopadhyay, A., et al. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-Ribose) polymerase inhibitors. Clinical Cancer Research. 16 (8), 2344-2351 (2010).
- Graeser, M., et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clinical Cancer Research. 16 (24), 6159-6168 (2010).
- Hill, S. J., et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discovery. 8 (11), 1404-1421 (2018).
- Naipal, K. A. T., et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clinical Cancer Research. 20 (18), 4816-4826 (2014).
- Tumiati, M., et al. A functional homologous recombination assay predicts primary chemotherapy response and long-term survival in ovarian cancer patients. Clinical Cancer Research. 24 (18), 4482-4493 (2018).
- Mukhopadhyay, A., et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Pesquisa do Câncer. 72 (22), 5675-5682 (2012).
- Johnson, S. W., Laub, P. B., Beesley, J. S., Ozols, R. F., Hamilton, T. C. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Pesquisa do Câncer. 57 (5), 850-856 (1997).
- Stefanou, D. T., et al. Aberrant DNA damage response pathways may predict the outcome of platinum chemotherapy in ovarian cancer. PLoS One. 10 (2), 0117654 (2015).
- Helleday, T., Petermann, E., Lundin, C., Hodgson, B., Sharma, R. A. DNA repair pathways as targets for cancer therapy. Nature Reviews Cancer. 8 (3), 193-204 (2008).
- Ivashkevich, A., Redon, C. E., Nakamura, A. J., Martin, R. F., Martin, O. A. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Letters. 327 (1-2), 123-133 (2012).
- Fuh, K., et al. Homologous recombination deficiency real-time clinical assays, ready or not. Gynecologic Oncology. 159 (3), 877-886 (2020).
- Cong, K., et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Molecular Cell. 81 (15), 3128-3144 (2021).
- Lee, E. K., Matulonis, U. A. PARP inhibitor resistance mechanisms and implications for post-progression combination therapies. Cancers. 12 (8), 2054 (2020).
- Panzarino, N. J., et al. Replication gaps underlie BRCA deficiency and therapy response. Pesquisa do Câncer. 81 (5), 1388-1397 (2021).
- Yee, C., Dickson, K. A., Muntasir, M. N., Ma, Y., Marsh, D. J. Three-dimensional modelling of ovarian cancer: from cell lines to organoids for discovery and personalized medicine. Frontiers in Bioengineering and Biotechnology. 10, 836984 (2022).
- Regan, J. L. Immunofluorescence staining of colorectal cancer patient-derived organoids. Methods Cell Biology. 171, 163-171 (2022).
- Graham, O., et al. Generation and culturing of high-grade serous ovarian cancer patient derived organoids. Journal of Visualized Experiments. (191), (2023).
- Jakl, L., et al. Validation of JCountPro software for efficient assessment of ionizing radiation-induced foci in human lymphocytes. International Journal of Radiation Biology. 92 (12), 766-773 (2016).
- Mullen, M. M., et al. GAS6/AXL inhibition enhances ovarian cancer sensitivity to chemotherapy and PARP inhibition through increased DNA damage and enhanced replication stress. Molecular Cancer Research. 20 (2), 265-279 (2022).
- van Ineveld, R. L., Ariese, H. C. R., Wehrens, E. J., Dekkers, J. F., Rios, A. C. Single-cell resolution three-dimensional imaging of intact organoids. Journal of Visualized Experiments. (160), e60709 (2020).
- Dekkers, J. F., et al. High-resolution 3D imaging of fixed and cleared organoids. Nature Protocols. 14 (6), 1756-1771 (2019).
- O’Rourke, K. P., Dow, L. E., Lowe, S. W. Immunofluorescent staining of mouse intestinal stem cells. Bio Protocols. 6 (4), 1732 (2016).
- Nwani, N. G., Sima, L. E., Nieves-Neira, W., Matei, D. Targeting the microenvironment in high grade serous ovarian cancer. Cancers. 10 (8), 266 (2018).
- Ghoneum, A., et al. Exploring the clinical value of tumor microenvironment in platinum-resistant ovarian cancer. Seminars in Cancer Biology. 77, 83-98 (2021).
- Yang, Y., Yang, Y., Yang, J., Zhao, X., Wei, X. Tumor microenvironment in ovarian cancer: function and therapeutic strategy. Frontiers in Cell and Developmental Biology. 8, 758 (2020).
- van de Wetering, M., et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 161 (4), 933-945 (2015).
- Broutier, L., et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nature Medicine. 23 (12), 1424-1435 (2017).
- Drost, J., Clevers, H. Organoids in cancer research. Nature Reviews Cancer. 18 (7), 407-418 (2018).
- Cybulla, E., Vindigni, A. Leveraging the replication stress response to optimize cancer therapy. Nature Reviews. , (2022).
