To keep the samples sterile, all steps involving parasite culture should be performed inside a biosafety level 2 (BSL-2) hood or according to local health and safety regulations. A graphical summary of this protocol can be found in Figure 1.
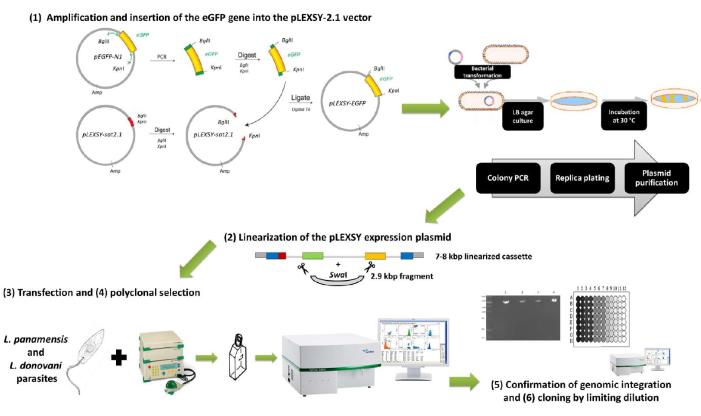
Figure 1: Summary scheme of the protocol for generating eGFP-expressing L. panamensis and L. donovani parasites using the pLEXSY-2.1 vector family. All six major sections described in the article are depicted here. 1) Amplification and insertion of the eGFP gene into the pLEXSY-2.1 vector: the target gene is amplified, adding the recognition sites for the enzymes KpnI and BglII, and both the eGFP amplicon and pLEXSY plasmid are sequentially digested with both enzymes for subsequent ligation with T4 ligase. 2) Linearization of the pLEXSY expression plasmid: the pLEXSY + eGFP construct is digested with SwaI for linearization and purification of the 7-8 kbp expression cassette. 3) Transfection and 4) polyclonal selection: Leishmania promastigotes are transfected with the expression cassette by electroporation and put back in culture for antibiotic selection of recombinant parasites. Parasite fluorescence is confirmed through flow cytometry. 5) Confirmation of genomic integration and 6) cloning by limiting dilution: integration of the pLEXSY-eGFP expression cassette is confirmed by diagnostic PCR, using a forward primer hybridizing to the expression cassette and a reverse primer hybridizing to a chromosomal sequence absent in the expression cassette. Transfected cultures could be enriched for fluorescent parasites by cloning by limiting dilution, and clones with the highest mean fluorescent intensities can be selected for further applications. Please click here to view a larger version of this figure.
1. Amplification and insertion of the eGFP gene into the pLEXSY-2.1 vector
- Amplification of the eGFP gene
NOTE: As this protocol uses the pLEXSY-2.1 vector family (see Table of Materials) for the constitutive expression of target proteins following the integration of the expression cassette into the chromosomal 18S rRNA locus (ssu) of Leishmania species, the first step is to introduce in the eGFP gene the sequences containing the restriction sites that allow its insertion into pLEXSY-2.1 vectors. In this case, as eGFP is only required to be expressed cytosolically, the restriction sites for the enzymes BglII and KpnI were added in the 5' and 3' ends of the eGFP gene, respectively. Cloning with KpnI results in the fusion of the target protein to a C-terminal polyhistidine tag of six residues, followed by a stop codon encoded in the pLEXSY-2.1 plasmid for further purification of the protein of interest. For this reason, the reverse primer sequence does not contain a stop codon.- Depending on the plasmid source for eGFP, analyze the target sequence using a restriction analysis tool such as NEBcutter (http://tools.neb.com/NEBcutter/index.php3)31 to make sure that it does not contain internal sites for the restriction enzymes used for cloning (BglII and KpnI) or for SwaI, which is the enzyme used for vector linearization prior to transfection.
- Design forward and reverse primers for eGFP amplification containing the BglII and KpnI restriction sequences. As an example, the eGFP source for this protocol was the pEGFP-N1-1X plasmid32, and the primer sequences were those reported by Bolhassani et al.27:
Forward primer (EGFP1): 5'-ATGATATCAAGATCTATGGTGAGCAAGGGC-3' (BglII restriction site in bold).
Reverse primer (EGFP2): 5'-GCTCTAGATTAGGTACCCTTGTACAGCTCGTC-3' (KpnI restriction site in bold). - Amplify the target gene using a high-fidelity polymerase to ensure the preservation of the coding sequence. As an example, for the primers EGFP1 and EGFP2, run the PCR cycling protocol as reported by Bolhassani et al.27. Run an initial denaturation of 2 min at 94 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 57 °C, and 1 min at 72 °C. Run a final extension step of 10 min at 72 °C. Add the primers at a final concentration of 0.2 µM.
- Purify the fragment using a conventional PCR product purification kit, or standard sodium acetate and ethanol precipitation, as described elsewhere33.
- Insertion of the eGFP into the pLEXSY-2.1 expression vector
- Trim the ends of the eGFP-PCR product using BglII and KpnI, following the manufacturer's instructions.
- First, set up the reaction for the enzyme requiring the lowest salt concentration (in this case, KpnI), according to the manufacturer's instructions, and incubate at 37 °C for 1 h.
- Then, add 100 mM NaCl and 10 units of BglII and incubate for 15 min. Purify the reaction, as described in step 1.1.4.
- Digest the pLEXSY expression vector with BglII and KpnI, following the sequential digestion protocol described in step 1.2.1.
- Ligate the pLEXSY vector and eGFP gene with T4 ligase using a molar ratio of 1:3 (vector:insert). Briefly add 100 ng of digested pLEXSY and 28 ng of digested eGFP product to a 20 µL reaction containing ligase buffer at a final concentration of 1x and 3 units of T4 DNA ligase. Incubate overnight at 4 °C.
- Transform competent E. coli cells, such as XL-10, DH5α, or DH10B, with the ligation product obtained in step 1.2.3 using standard transformation protocols33. Plate the transformed cells in Luira-Bertani (LB)-ampicillin agar and incubate for 24 h at 30 °C to select recombinant clones (pLEXSY plasmids are more stable in E. coli at 30 °C than at 37 °C).
- Screen for the presence of the insert in the plasmids by colony PCR.
- Pick individual colonies, resuspend in 20 µL of nuclease-free water, heat at 95 °C for 15 min, and take 1 µL of the lysate as a DNA template for colony PCR. Use the forward primer for eGFP amplification, described in step 1.1.2 (in this example, EGFP1), and the primer A264 (5'-CATCTATAGAGAAGTACACGTAAAAG-3') as a reverse primer29.
- The primer A264 is designed to anneal 80 bp after the stop codon of the insert in pLEXSY-2.1 expression vectors. As an example, for the primer pair EGFP1 + A264, use a PCR cycling protocol consisting of an initial denaturation of 2 min at 94 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 50 °C, and 1 min at 72 °C. Run a final extension step of 10 min at 72 °C. The primers are added at a final concentration of 0.2 µM.
NOTE: The expected product using this primer set is 859 bp long. The annealing sites for the primers EGFP1 and A264 are depicted in Figure 2.
- Prepare the purified plasmid DNA from a positive clone for subsequent transfection using a commercial plasmid isolation kit or standard alkaline precipitation33. Use 50 mL of an overnight culture at 30 °C for isolation of a minimum of 10 µg of plasmid/positive clone.
- Trim the ends of the eGFP-PCR product using BglII and KpnI, following the manufacturer's instructions.
2. Linearization of the pLEXSY expression plasmid
- Use at least 10 µg of the purified pLEXSY-eGFP plasmid for digestion with SwaI (recognition site: 5'-ATTTAAAT-3'). Digest for 3-4 h at 25 °C and heat-inactivate at 65 °C for 20 min.
- Run the digestion product in agarose gel electrophoresis to separate the resulting fragments. This digestion will generate two fragments, a 2.9 kbp fragment representing the elements necessary for replication and selection in E. coli, and a 7-8 kbp fragment representing the linearized expression cassette.
- Purify the expression cassette using a commercial agarose gel extraction kit.
3. Transfection of L. panamensis and L. donovani by electroporation
NOTE: Leishmania culture is performed as described elsewhere34. In this example, L. panamensis and L. donovani promastigotes were cultured in Schneider's insect medium, supplemented with 20% (v/v) fetal bovine serum (FBS) and 50 µg/mL gentamicin, and incubated at 26 °C.
- Grow the parasite cultures until there are enough log-phase or early stationary-phase promastigotes to use approximately 4 x 107 parasites per transfection. The maximum cell density per culture flask before reaching the stationary phase may vary depending on the Leishmania species and how well the strains are adapted to laboratory conditions. Pool the contents of multiple culture flasks if required.
- Centrifuge the parasite culture at 2,000 x g for 3 min at room temperature (RT).
- Resuspend the pellet in electroporation buffer at 4 °C (21 mM HEPES, 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, and 6 mM glucose; pH 7.5)27 to get a concentration of 1 x 108 parasites/mL. Put on ice for 10 min.
NOTE: The electroporation buffer must be sterilized by filtering before use. - In parallel, pre-chill tubes with 2-10 µg of linearized pLEXSY-eGFP construct in a maximum volume of 50 µL of water or 10 mM tris buffer (pH 8.0) and electroporation cuvettes of d = 2 mm.
- Add 350 µL of pre-chilled parasites to the tube with the linearized plasmid and transfer the entire 400 µL to the electroporation cuvette on ice. In parallel, electroporate the parasite cells without plasmid DNA as a negative control.
- Electroporate using one of these two protocols:
Exponential decay: 450 V, 450 µF, one pulse.
Time constant: 450 V, T = 3.5 ms, one pulse.
NOTE: These protocols were run using a commercial gene pulser (Table of Materials) with PC and CE modules. - Put the cuvette back on ice for 10 min and transfer the electroporated parasites to 5 mL of the appropriate culture medium. This example used Schneider's insect medium supplemented with 20% (v/v) FBS. Incubate at 26 °C for 20 h.
4. Polyclonal selection in culture
- Approximately 20 h post-electroporation, observe the cultures under a microscope. At least half of the parasite population should show visually good morphology and motility (drop-like promastigotes with oscillating flagellum moving through the media and/or forming active parasite aggregates). Add the appropriate selective antibiotic depending on the pLEXSY plasmid used. In this example, pLEXSY-sat2.1 is used, which contains streptothricine acetyltransferase as a selection marker. Therefore, use nourseothricin as a selective antibiotic at a 0.1 mg/mL concentration.
- Follow the cultures microscopically until a clear difference between the parasites electroporated with and without plasmid DNA is seen.
- Verify the parasite fluorescence through flow cytometry.
- Centrifuge 1 mL of the stationary-phase culture at 2,000 x g for 3 min at RT. Wash twice in PBS and resuspend the final pellet in 1 mL of PBS.
NOTE: The fluorescence of eGFP can be detected in the FL1 channel of most commercial cytometers. Gain settings for forward and side scatter, and the FL1 channel may vary between cytometers. For this example, a CyFlow space (Sysmex) cytometer was used. - Run the samples, collecting 20,000 events at a speed of 0.5 µL/s, and set the gain values for the forward scatter (FSC), side scatter (SSC), and fluorescence 1 (FL-1) channels as 225.0, 200.0, and 520.0, respectively. Run a control sample with non-fluorescent parasites to determine the parasite population in a FSC versus SSC dot-plot.
- Create a gate (G1) containing the parasite population and filter the FL-1 channel through that gate for determining the autofluorescence of promastigotes and setting a range gate (G2) for the FL-1 channel.
- Run the transfected parasites to verify if there is fluorescence. Record the percentage of parasites in G1 that are fluorescent and the mean of fluorescence intensity in G2.
- Centrifuge 1 mL of the stationary-phase culture at 2,000 x g for 3 min at RT. Wash twice in PBS and resuspend the final pellet in 1 mL of PBS.
5. Confirmation of genomic integration
NOTE: Integration of the pLEXSY-eGFP expression cassette can be confirmed by diagnostic PCR, using a forward primer hybridizing to the expression cassette (varies depending on the pLEXSY-2.1 vector used) and the reverse primer F3002 (5'-CTGCAGGTTCACCTACAGCTAC-3') hybridizing to a chromosomal ssu-flanking sequence absent in the expression cassette. In this example, the F2999 forward primer (5'-CCTAGTATGAAGATTTCGGTGATC-3') is used as it hybridizes in the pLEXSY-sat2.1 expression cassette. A schematic representation of the integrated expression cassette and primer annealing sites for diagnostic PCR is depicted in Figure 2.
- Purify the genomic DNA from 2-5 mL of a stationary-phase parasite culture by conventional phenol/chloroform extraction35 or with a commercial kit.
- Perform the diagnostic PCR for pLEXSY-sat2.1 using a cycling protocol consisting of an initial denaturation of 2 min at 94 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 53 °C, and 1 min at 72 °C. Run a final extension step of 10 min at 72 °C. The primers are added at a final concentration of 0.2 µM.
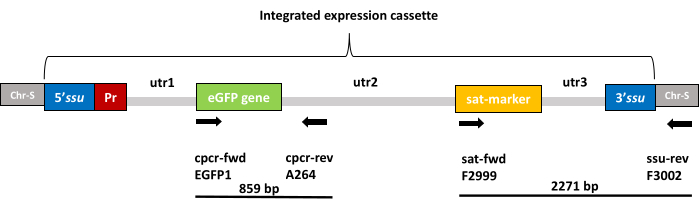
Figure 2: Schematic representation of the integrated expression cassette. Boxes labeled as Chr-S (gray) represent the adjacent chromosomal sequences of the 18S rRNA locus (ssu) into which the expression cassette is integrated. The integrated expression cassette consists of 5' and 3' sequences (blue) for homologous recombination into the ssu locus, an additional Leishmania ribosomal promoter (red) for enhanced protein production, the eGFP gene (green), a selection marker gene (yellow), and three untranslated regions (light gray), namely utr1, 2, and 3, which provide the splicing signals for post-transcriptional mRNA processing for optimized expression of eGFP and the selection marker in Leishmania parasites. Annealing sites for forward and reverse primers used for verification of the insert presence by colony PCR (step 1.2.5) are marked by the black arrows labeled as cpcr-fwd (EGFP1 primer) and cpcr-rev (A264 primer) respectively, which delimit an 859 bp product. The annealing sites for forward and reverse primers used for the verification of genomic integration by diagnostic PCR (step 5) are marked by the black arrows labeled as sat-fwd (F2999 primer) and ssu-rev (F3002 primer), respectively, which delimit a 2,271 bp product. Please click here to view a larger version of this figure.
6. Cloning by limiting dilution (optional)
- After confirmation of fluorescence through flow cytometry, prepare a dilution of recombinant parasites at a concentration of five promastigotes/mL36.
- Add 100 µL of this dilution into each well of a 96-well plate. In this manner, the plates are seeded at an average density of 0.5 promastigotes/well, which ensures that some wells receive a single parasite while minimizing the probability of having more than one parasite per well36.
- Leave the plate undisturbed for at least 12 h. Follow the plate microscopically at a minimum magnification of 100x until wells with parasite growth are detected.
- When a plate well is full of parasites, use the content to inoculate a 5 mL culture. This takes 1-2 weeks.
- When clone-seeded cultures reach the stationary phase, verify fluorescence using flow cytometry, as described in step 4.3. Select the clones to be used for further in vitro and in vivo assays based on the percentage of fluorescent parasites and the mean fluorescence intensity measured in the FL-1 channel. Aim for clones with 98%-99% of fluorescent parasites and select those with the highest mean fluorescence intensity.
After building the pLEXSY-eGFP construct and transforming competent E. coli cells, colonies containing the construct with the eGFP insert will generate an approximately 859 bp product after running the colony PCR described in section 1.2 (Figure 3A). Total digestion of the purified plasmid from positive colonies using SwaI should give two characteristic fragments in gel electrophoresis, a 2.9 kbp fragment that is the portion of the PLEXSY vector containing all the necessary elements for replication and selection in E. coli, and a 7-8 kpb fragment representing the linearized expression cassette that is used for transfection (Figure 3B).
Following transfection, the polyclonal selection is made mostly qualitatively. The status of the parasite cultures during selection with antibiotics is monitored microscopically, bringing special attention to parasite morphology and motility. After successful recovery of a culture of transfected parasites, those effectively expressing eGFP should be detected in the FL-1 channel of a flow cytometer (Figure 4A); transfection efficiencies may vary between different Leishmania species. In this work, after polyclonal selection, 98.44% of L. donovani parasites analyzed by flow cytometry were fluorescent in comparison with 82.00% of L. panamensis (Figure 4B). If the pLEXSY-eGFP expression cassette has been successfully integrated into the ssu locus, a 2.3 kbp product should be observed after running the diagnostic PCR described in section 5 (Figure 4C). These results ensure that recombinant parasites are obtained that constitutively express eGFP as an integrated transgene and will remain stable in subsequent passages of the parasite culture.
Cloning by limiting dilution allows the enrichment of the fluorescent parasite population with lower transfection efficiencies, such as L. panamensis, as well as the selection of clones with the highest fluorescence intensities. Four clones were obtained for each of the Leishmania species used in this work (Table 1), namely Lpan-A7, F11, F12, and H2 for L. panamensis, and Ldon-C1, G4, F2, and H1 for L. donovani. Clones with the highest percentage of fluorescent parasites (Lpan-F11 = 95.74%; Ldon-C1 = 99.16%) and mean fluorescence intensity (Lpan-F11 = 35.63 relative fluorescent units [RFU]; Ldon-C1 = 14.12 RFU) were selected for further use in drug screening assays.
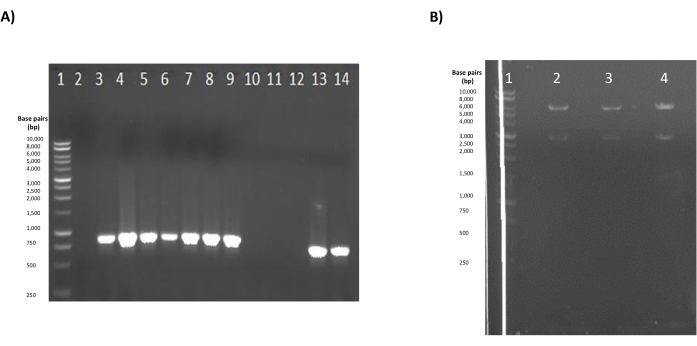
Figure 3: Representative results of preparation of the pLEXSY-eGFP construct for transfection. (A) Representative results of the colony PCR. Lane 1: 1 kb ladder (units in base pairs); lanes 2, 10, and 12: negative samples for the presence of the eGFP insert; lanes 3-9 and 13-14: positive samples for the presence of the eGFP insert. (B) Results of the linearization with SwaI. Lane 1: 1 kb ladder (units in base pairs); lanes 2-4: digestion reactions of plasmids purified from three different colonies positive for the presence of the eGFP insert. A 2.9 kbp fragment representing the elements necessary for replication and selection in E. coli, and a 7-8 kpb fragment representing the linearized expression cassette are observed. Both gels were prepared at an agarose concentration of 1%. Please click here to view a larger version of this figure.

Figure 4: Representative results of confirmation of eGFP expression and chromosomal insertion of the expression cassette. (A) Flow cytometry histogram in the FL-1 channel. Red: wild-type cells without eGFP; Blue: transfected L. panamensis parasites with 82.00% of the population expressing eGFP. (B) Comparison of transfection results between L. panamensis (Lpan-1) and L. donovani (Ldon-1). A higher transfection efficiency was observed in L. donovani (98.44%) than L. panamensis (82.00%) after polyclonal selection. (C) Results of the PCR for confirmation of genomic integration. L: 100 bp ladder (units in base pairs); lanes 1-4: samples from four different clones of L. panamensis showing a characteristic 2.3 kbp product for pLEXSY-sat2.1 integration into the ssu locus. Please click here to view a larger version of this figure.
| Species | Clone code | Percentage of fluorescent parasites | Mean fluorescence intensity* |
| L. panamensis | Lpan-A7 | 90.00 | 24.70 |
| Lpan-F11 | 95.74 | 35.63 | |
| Lpan-F12 | 92.85 | 34.44 | |
| Lpan-H2 | 85.00 | 20.66 | |
| L. donovani | Ldon-C1 | 99.16 | 14.12 |
| Ldon-G4 | 99.21 | 13.96 | |
| Ldon-F2 | 97.96 | 11.67 | |
| Ldon-H1 | 98.97 | 12.90 |
Table 1: Clones obtained by limiting dilution. Four clones were obtained by limiting dilution for each of the Leishmania species used in this work. Percentage of fluorescent parasites and mean fluorescence intensity are reported for each clone. *Fluorescence is expressed in relative fluorescence units (RFU).
