Transient absorption (TA) spectroscopy is a time-resolved spectroscopic technique that monitors the evolution of photo-excited species through time-dependent changes to their absorption spectrum following excitation with a pulse of light. Because TA is an absorptive technique, spectroscopic signals that arise from states that undergo both radiative transitions (i.e., states that typically emit a photon) and non-radiative transitions (states that are typically non-fluorescent and undergo internal conversion, intersystem crossing or participate in photoreactions) can be identified and their evolution followed1,2. Depending upon the specifics of the excitation source and detection method, TA allows access to kinetics from femtoseconds to beyond microseconds and from the UV to the far IR, making it a versatile spectroscopic tool. Commercialization of TA spectrometers has advanced significantly over the past several decades, leading to more laboratories and facilities having access to this powerful technique2.
Modern TA systems are capable of producing large datasets with high energetic and temporal resolution. The datasets generally take the form of a 2D matrix of transmittance or absorbance difference values as a function of wavelength and time delay relative to the excitation pulse. This dataset can be viewed as a two-dimensional heat map or a three-dimensional topographical map. Interpretation of these data has become more complex as researchers strive to include the entire dataset when generating fits that best describe their system of interest3.
Although TA can cover a broad range of wavelengths and time scales, this protocol focuses on one of its most widely accessible forms4: broadband spectroscopy in the UV-visible region driven by a femtosecond pulsed laser. A schematic5,6 of such an instrument is provided in Figure 1. The experiment begins by taking a pulse from the laser and splitting it into two copies. One copy of the pulse, called the 'pump,' is used to excite the sample. A device such as an optical parametric amplifier (OPA) is typically used to transform the pump pulse to the desired excitation wavelength5,7. The second copy of the pulse, called the 'probe,' enters a mechanical delay stage, which can vary the time delay between the pump and probe pulses by varying the distance the pulse travels. The single-wavelength probe pulse is then transformed into a white-light continuum using a sapphire or calcium fluoride (CaF2) crystal8. The white-light pulse is passed through the sample, and its spectrum is measured using a broadband detector such as a charge-coupled device (CCD) camera. By measuring changes in the spectrum of the white-light pulse with and without the pump, changes in the sample's absorption spectrum induced by the pump, ΔA(T), can be measured. Interested readers are directed toward this useful review9 for more information about the detection process.
In all forms of TA spectroscopy, the ΔA(t) spectra are calculated by taking the difference between the ground-state absorption, Aprobe, and that of the excited state, Apump+probe, at a given time delay, t, between the two pulses2,5,9,10.
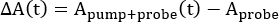 (1)
(1)
Note that Aprobe is equivalent to the sample steady-state absorption spectrum and is time-independent; the experiment's time resolution arises from the delay between the pump and probe captured in Apump+probe(t). A simulation of these data is shown in Figure 2A.
In contrast to steady-state absorption spectra, TA spectra can have both positive and negative features due to the difference taken in Equation 1. Positive features are the result of new absorbing species created by the pump pulse and can represent excited chromophore states, triplet states, geometric rearrangements, solvation effects, or excited-state photoproducts3. General guidelines for identifying these features and assigning them to chemical species will be presented in the Discussion. Negative features can arise from either the ground-state bleach (GSB) or stimulated emission (SE) (Figure 2B). The GSB is due to the loss of ground-state population following absorption of the pump pulse. Molecules promoted to the excited state no longer absorb in the same region as their ground state; therefore, less of the probe pulse is absorbed, and the difference in Equation 1 can be negative in that region. The GSB is characterized by having the same spectral shape as that of the ground-state absorption but with an opposite sign. SE signals result from emission from an excited-state species stimulated by the probe pulse3. Emission from these species results in more light reaching the detector, which is equivalent to having less absorption at those wavelengths. The SE signal will have a similar spectral shape as the spontaneous emission spectrum of the species, but with a negative sign and a different frequency weighting10.
In addition to information about excited-state species, TA spectra can contain a number of artifacts and extraneous features that can distort the underlying dynamics and obscure the assignment of absorption bands11. Improper treatment of these artifacts in the data preparation and analysis can lead to the application of inappropriate photophysical models to the data and, consequently, to misleading conclusions11. Therefore, the first part of this protocol focuses on how to properly process TA datasets after they are collected. The goal of this section is to provide researchers new to TA with a set of guidelines that will help develop an intuition and appreciation for the rigorous preparation and processing of their data.
After a dataset is processed, a plethora of tools and models are available for fitting and interpreting the spectra with various levels of complexity and rigor10. The goal of the second section of this protocol is to prepare the reader to apply single-wavelength fitting and global analysis to data and to provide guidance as to when these models are appropriate for describing their data. Commercial software is now readily available for preparing and treating TA data, such as Surface Xplorer12,13 from Ultrafast systems (free to download and use, see Table of Materials). Other free alternatives have been released by academic researchers, such as Glotaran14. Glotaran is a free software program developed for global and target analysis of time-resolved spectroscopy and microscopy data. It serves as a graphical user interface (GUI) for the R-package TIMP14. Additionally, users can use programming languages like Python to write their own codes that carry out the analysis. Each of these fitting software and programming solutions have positive features that make them important contributions. For the purpose of this study, we can present only one software for the visual component of this activity. An in-depth discussion of each fitting software is beyond the scope of this article.
This article provides a step-by-step procedure for (1) processing TA data, (2) fitting TA data using single-wavelength kinetics and global analysis, and (3) extracting data and fitting it to other models. A set of representative TA data is included for the reader to use as practice (Supplementary File 1 and Supplementary File 2). The data is a measurement of a 165 µM sample of 1, 4-bis(5-phenyloxazole-2-yl)benzene (POPOP) in ethanol excited at 330 nm and collected over a range of −5 ps to 5.5 ns. Additionally, a "blank" sample containing ethanol only and no sample was collected under the same experimental conditions over a range of −5 ps to 5 ps, which is used in preparing the data for fitting (step 1). Spectra were collected using an ultrafast transient absorption spectrometer. The sample was contained in a 2-mm pathlength cuvette and subjected to constant stirring. The processing and fitting procedure described is based on the Surface Xplorer software that fits data in the *.ufs format, and that will herein be called the "fitting program". Programs for converting datasets in other formats to *.ufs files are available15. Although the details of this protocol are specific to Surface Xplorer, the steps that follow are generalizable to any software package, commercial or home-built. Additionally, the results of data processing may be extracted and fit using these other software packages. A supporting information file (Supplementary File 3) provides additional advice on fitting.
1. Preparing data for fitting
- Load the SAMPLE dataset into the Fitting software. Data will appear as shown in Supplementary Figure 1.
- When scattered excitation light is present within the optical detection window of the experiment, use the Subtract Scattered Light option (Supplementary Figure 2). If no scattered excitation light is present in the data, proceed to step 1.5.
NOTE: Scattered light is most commonly observed when the excitation wavelength falls within the optical window. Scattered light appears as a sharp negative (bleach) feature at the excitation wavelength (or some diffraction order, or wavelength produced in the OPA) that does not vary with time. - Click on the Surface menu, then click on the Subtract Scattered Light option (Supplementary Figure 2). A new window will appear.
- In the new window, click the arrow buttons to set the number of background spectra to average (Supplementary Figure 3) in order to perform background correction. Using ten spectra provides a good starting point and the number can be adjusted as desired. Click on Accept to perform the subtraction (proceed to step 1.7).
NOTE: Background spectra are drawn from the first spectra present in the dataset and then move forward temporally using as many background spectra as needed to provide averaging of the background signal; however, using too many will begin to use spectra that contain the signal of interest so do not use too many. With long time-window data, the scattering feature may not appear at the end of the time window of data. This might occur if the time window exceeds the camera integration time, or other reasons, based on how the TA experiment is constructed. To correct this, the set time range option can be used as outlined in Supplementary File 3. - For data where there is no scattered light present within the optical window, click on the Surface menu, then click on the Subtract Background option.
- In the window that appears, click the arrow buttons in the lower right for "Number of Spectra" to average (choose 10), and click on Accept.
NOTE: Background spectra in this option function the same as they do in the "Subtract Scattered Light" option. Starting with ten spectra provides good averaging. More spectra can be used, but care must be taken not to use too many so as to avoid including spectra containing signals of interest. The user manual for Surface Xplorer describes the differences in the correction applied for the "Subtract Scattered Light" vs. the more basic "Subtract Background" correction. It is important to apply the appropriate correction for the artifact16. - Data near the edges of the optical window may have a very low signal-to-noise ratio resulting from the shape of the probe spectrum and/or the sample absorbing too much of the white light. Noisy data in these regions makes analysis more difficult. Remove these unhelpful portions of the spectrum. Click on the end wavelengths on the spectrum (lower left tile), type in new values (Supplementary Figure 4), and click on enter. Choose a wavelength range that removes the noisy data at the edges of the window. For the provided data, the range is 340-680 nm.
- Finalize the adjustment of the spectral window to the desired range of wavelengths. Click on the Surface menu, then click on Crop (Supplementary Figure 5). A pop-up window will appear.
- Click on OK. Click on the File menu, then click on Save File As. Then, click on OK. Close this dataset.
NOTE: Be careful when cropping, as the function will crop data along both the wavelength and time delay axes, deleting all other data. Ensure that the time delay window contains the portion of the data to be preserved. Also, it is strongly recommended to save any cropped data as a new, appropriately labeled file so as to leave the raw data surface intact as a backup.
- Click on OK. Click on the File menu, then click on Save File As. Then, click on OK. Close this dataset.
- For data collected on the fs or ps time scales, a chirp correction must be applied. Open the data surface with only the solvent or substrate (no sample) taken in the same experimental set up as the data. This sample is called the "blank" experiment run. Perform the same set of steps on the "blank" data (from step 1.2 to step 1.8) that were performed on the sample data.
NOTE: It is strongly recommended that such a "blank" experiment be run under the same conditions as the sample but with a shorter time window (for example, ~ −5 ps to 5 ps) to ensure a high number of points around time zero. This "blank" run should consist of only solvent or substrate, depending on the sample type, and is used to establish the chirp curvature. Preparation of the "blank" up to this point should follow the same background correction and cropping as the sample data. If a "blank" was not run, the chirp correction can be done directly on the dataset. - Start the chirp correction process. In the heat map tile (top left), click on the crosshair and drag the vertical line component to the blue end of the spectrum. Start on the blue wavelength range near the start of the spectral window. Click on the Kinetics menu, then click on Fit Solvent Response.
NOTE: Fit solvent response should only be performed on a "blank" sample that does not generate any signal past time zero. Attempting to apply this fit function to a dataset containing data on a molecule or material of interest will result in the program attempting to fit the data instead of the IRF (instrument response function). For "blank" samples, the only signal present should be a coherent artifact arising from cross-phase modulation. Cross-phase modulation occurs only where the pump and probe beams overlap and, therefore, provides a trace of the chirp curvature that can be fit using the "Fit Solvent Response" option. Manual placing of points will be required for data without an accompanying "blank" to use for correction and are described in more detail in Supplementary File 3. - A new "Fit solvent response" window opens. Click on the Fit button (Supplementary Figure 6). The fitting will generate a fit to the instrument response function using the first and second derivatives of a Gaussian. Click on the Save button, then click on x to close the screen.
NOTE: The red fitted line should match the data points (blue hollow squares) well across the entire time range, most importantly the large feature seen near time zero (0.1-2.0 ps). The fit will have the most success if there are a high number of points around time zero, which can be achieved by using a short time delay window for the "blank" experiment and retaining a large number of points. If the fit does not seem to match the data points well, check the "Add Gaussian (R0)" box and retry the fit. This option will add a sum of the Gaussian to the first and second derivatives and may better fit the IRF feature shape at this wavelength. If the solvent response fitting still fails to capture the IRF signal, select a different wavelength. - Perform this process (steps 1.10-1.11) at least five times, as five points will be required to properly correct for a chirp. The points should be spaced out across the entire spectral window if possible. Some solvents/substrates may not generate an observable signal in portions of the spectral window, depending on experimental conditions. More points may be added/used as needed to produce an acceptable fit. When complete, close the "blank" dataset.
NOTE: Each saved point is written to an Excel file in the working folder as a new row as soon as the user clicks on Save. If an undesired point is saved, the point can be removed from the Excel file by deleting the undesired point's row. - Re-open the cropped and background subtracted dataset. Click on the Surface menu, then click on the Chirp Correction option. This will bring up a new screen with three windows and a menu in the bottom right (Supplementary Figure 7).
- Add the chirp correction that was just created. Click on Add from file option, select the Excel file that ends in "fit coefficients", and click on OK. The chirp correction fit is now displayed on the upper left window as a black line with X markers (Supplementary Figure 8).
NOTE: The chirp correction is displayed as a solid line; the X markers along the line are the points generated from the fit solvent response process. Points can be added manually by adjusting the crosshairs and clicking on Add. Points can also be removed by highlighting them and pressing Remove. Further points can be hand-edited by typing in values in the list at the bottom right. Finally, if desired, the current correction can also be saved as a file for later reuse using the Save to file button. - Click on the Preview chirp correction button. This temporarily applies the chirp correction. Observe the correction in the top left window to ensure that the data has been temporally flattened and no more curvature is observable.
- If satisfied with the chirp correction, click on the Apply & Exit button. If not satisfied, repeat steps 1.10-1.14, choosing more (or different) wavelengths for the chirp correction fit until a satisfactory correction is obtained.
NOTE: Applying the chirp correction will adjust the time zero to the straightened line as it appears in the preview. There may be some temporal misalignment between the "blank" and the data surface.
- If satisfied with the chirp correction, click on the Apply & Exit button. If not satisfied, repeat steps 1.10-1.14, choosing more (or different) wavelengths for the chirp correction fit until a satisfactory correction is obtained.
- Click on the File menu, then click on Save File As. Type an appropriate name for the file to indicate a chirp correction has been applied. Then, click on OK.
- Some scattering features in the data may not be completely removed when performing the background subtraction. These features impact fitting and produce erroneous fitting results. Locate any such features in the data that should be removed. The scattering feature can be most easily identified in the negative time region.
- In the upper left heat map tile, click and drag the crosshair to the negative time region. Staying within the negative time region, use the crosshair to determine the wavelengths at which the scattering feature begins and ends. Note the wavelength range of the scattering feature (for the provided dataset, the feature range is 654 nm to 672 nm).
NOTE: When determining if a feature should be removed, drag the horizontal crosshair up and down through the time axes to visualize the spectral range of the feature. Scattering features typically have very noisy single-wavelength kinetic traces, so the spectral range of the feature may also be verified using the kinetic traces.
- In the upper left heat map tile, click and drag the crosshair to the negative time region. Staying within the negative time region, use the crosshair to determine the wavelengths at which the scattering feature begins and ends. Note the wavelength range of the scattering feature (for the provided dataset, the feature range is 654 nm to 672 nm).
- Starting with the lower (blue) wavelength (i.e., 654 nm), click on the right-end wavelength on the spectrum (lower left tile) and type in the value of the lower (blue) feature extent.
- Crop the data by clicking on the Surface menu, then click on Crop. Click on OK in the popup menu. Save the cropped data with a unique file name to indicate which side of the data this is (Blue or Left is recommended). Close the file.
- Open the file with the applied chirp correction saved in step 1.16. Continue to the higher (red) wavelength extent of the feature. Click on the left-end wavelength on the spectrum (lower left tile) and type in the value of the higher extent of the feature.
- Crop the data by clicking on the Surface menu, then click on Crop. Click on OK in the popup menu. Save the cropped data with a unique file name to indicate which side of the data this is (Red or Right is recommended).
- Combine the two files by clicking on the File menu, then click on Combine Multiple Surfaces. In the new window, select both sides of the data (i.e., Right and Left or Blue and Red). Use ctrl + click to select each file. Verify that both files are selected in the "File Name:" box, then click on OK in the bottom right. When the progress bar finishes, the data has been combined.
NOTE: Any number of files can be combined in this fashion. Data can be stitched together across multiple cuts in both the time and wavelength axes. - Click on the File menu, then click on Save File As and choose a unique file name to indicate it is combined (Combined or Composite recommended). Then, click on OK to save the file.
NOTE: See section 3 for information on how to save data from the raw data window for later display and plotting. The data in the heat map (upper left tile) should appear as in Figure 3 and is now ready to be fit. Visualizing the representative spectra, as shown in Figure 3, is described in step 3.1.2.
2. Performing a Fit
- Load the properly prepared data surface.
- Decide which fitting will be performed and move to the appropriate section.
NOTE: This protocol presents two data fitting options: step 2.3 presents single-wavelength kinetic trace fitting, and step 2.4 presents global analysis fitting. - Single wavelength fitting
- To set up a single kinetic fit, move the cursor (either in the top or bottom left tiles) to the desired wavelength. Click on the Kinetics menu, then click on Fit Kinetic. For the provided dataset, start with 632 nm.
- In the new window that opens (Supplementary Figure 9), observe that the main fitting parameters and seed values are set in the upper left of the window underneath the program logo in the box next to this area under "current fit @ wavelength" text.
- Click on the arrow buttons to adjust the number of lifetimes (i.e., the number of exponential decays used to fit the data) in the "Finite lifetimes" box. For the provided dataset, choose 2 lifetimes. One to 3 lifetimes are typical as a starting point.
- If the data signal extends beyond the collected time window, an "infinite" lifetime component should be included. To do this, click on Use infinite lifetime checkbox. If the data fully decays to the baseline, do not check this box. For the provided dataset, do not check the box.
NOTE: The "infinite lifetime" allows a signal offset to remain (i.e., the program will not force the fit to decay back to the baseline). The use of an infinite component is required when the signal at that wavelength does not decay to the baseline within the time range of the experiment. - Enter guess values for the lifetimes and associated amplitudes, instrument response time, and time zero to help the fitting process (Supplementary Figure 10). Click on the desired parameter. Click in the value window, type in a guess value, and then click the initial guess button to set the value. For the provided dataset, appropriate guess values are: 0 = 0 ps, IRF = 0.25 ps, A1 = 0.6, t1 = 100 ps, A2 = 0.08, t2 = 1100 ps.
NOTE: "0" is the estimate of time zero, "IRF" is the instrument response time, "A" refers to the amplitude of a given exponential (see Equation 3), and "t" is the lifetime/time constant. Providing good guess values helps the program obtain a reasonable fit. Choose "A" values that are within the values of the A range in the dataset. Choose "t" values on time ranges where significant change is observed in the kinetic trace. The best way to gain intuition about how guess values affect the fit is to try several sets of guess values and observe the fits they produce. If one or more of these parameters are known, that parameter can be set and "fixed" so that it does not vary with the fit (Supplementary Figure 11). - When all guess parameters are entered, click on the Fit button. A representative fit is shown in Figure 4.
NOTE: Applying the fit will populate the data plot with the fit line and a residual plot that can be used to assess the quality of the fit. Fit parameters, such as the lifetimes and associated amplitudes, time zero, and instrument response time, are also populated in the upper left box. Use several different fit parameters to determine the number of lifetimes and the inclusion/exclusion of an "infinite" time component that produces the best fit to the data. - Save the fit by clicking on the Save button (Supplementary Figure 9).
NOTE: See section 3 for information on how to save data from the raw data window for later display and plotting.
- Global analysis fitting
- Click on the Surface menu, then click on the Principal Components via SVD option. A new window appears (Supplementary Figure 12).
NOTE: The top right window displays the principal kinetic traces, and the bottom left displays the principal spectra. The upper left tile displays a plot of the residual surface created by the difference between the original surface and a surface created by the selected principal components.
plot of the residual surface created by the difference between the original surface and a surface created by the selected principal components. - Click on the arrow buttons to set the "Number of Principal Components" (Supplementary Figure 12). For the provided dataset, choose 15.
NOTE: When deciding on the number of principal components to choose, one way is to keep increasing the number until both the principal spectra and principal kinetic traces resemble a noise pattern. Another way to determine how many principal components to choose is by looking at the weight coefficient values that appear to the left of the legend in the top right tile. Continue adding principal components until this value reaches 0.01. Generally, it is advisable to add a few more beyond this for good measure. This may result in as many as 15 or more principal components being selected. - Click on the Save button. Saved principal components are required to advance to the next step.
NOTE: Each principal component is a reduced complexity representation of the original data surface. Using principal components will result in a simplified surface compared to the raw data being analyzed. Accounting for most of the major features of the data surface is very important for getting an accurate fit, so it is key to use enough principal components to capture those features. Using more principal components will not compromise the fit. Therefore, if there is any doubt about how many principal components to select, use more principal components rather than fewer. Keep in mind that using too many might slow the fitting software. - After saving the principal components, the program will return to the main screen and global fitting can now be attempted. Click on the Surface menu, then click on the Global Fit option. A new window will open (Supplementary Figure 13).
NOTE: Principal kinetic traces are displayed in the upper right tile. The upper left tile will display a surface of the fit surface compared with the raw surface. The lower left tile will display the decay-associated difference spectra (DADS) generated by the fit. Finally, the bottom right tile is where the fit parameters can be set, including the number of exponential functions to use and whether to use an infinite function. - Use the arrow buttons next to the "number of exp." to set the number of exponential functions to include in the fit. If the data signal extends beyond the collected time window, an "infinite" lifetime component should be included. To do this, click on Use offset (Ainf) checkbox. For the provided dataset, choose 2 and do not click the box. If the data fully decays to the baseline, do not check this box.
NOTE: Fit parameters can be fixed before performing a fit by clicking on the label column in the bottom right box under global fit coefficients. The label will turn red and change to have a (fixed) indicator on the label. Any value typed in the box to the right will be used for that parameter rather than freely varied for the fit. Care should be taken when fixing values for a fit, as this may bias the fit results. - Click on the Fit button. Fit progress will be displayed via a small loading bar in the center of the screen. Upon completion of the fit progress, the windows will be populated with data from the fit (Figure 5 and Supplementary Figure 14). Visually examine the fitting results.
NOTE: Information from both the principal kinetic fit and DADS are used to determine if the fit is worth saving or is too poor. Generally, if the fit of the principal traces matches well with the data, and there are no or very few features in the plot, the fit can be accepted. It is easy to check multiple fits by changing the number of lifetimes and/or checking/unchecking the "Use offset (Ainf)" button. The best fit produced after checking multiple variations of the fitting parameters should be accepted. - Click on the Save button. This will save the currently shown fit along with the data in an Excel file.
NOTE: The Excel file is saved in the same file location as the dataset. If additional fits are performed and need to be saved, they will overwrite any previous version. As such, before generating and saving a new fit, give any old fit a unique name. Parameters saved from the fit include only the time zero, IRF, lifetimes and their accompanying DADS. This file does not contain any information about the plot or the fitting to the principal kinetic traces. Saving the principal kinetics traces is outlined in step 3.3. See step 3.0 for information on how to save data from the raw data window for later display and plotting.
- Click on the Surface menu, then click on the Principal Components via SVD option. A new window appears (Supplementary Figure 12).
3. Extracting raw data and fits from the fitting software for plotting
NOTE: Raw data or fits produced from single wavelength fitting or global analysis can be exported to csv files that can be opened in a range of other programs.
- Extracting raw data for plotting
- To export the heat map of the dataset, click on the File menu, then click on Export to CSV (Supplementary Figure 15). This will open a window, click on OK to save the csv file in the same directory as the data file open with the same name as the data file.
NOTE: Alternatively, raw data can be exported by right-clicking on the heat map window and clicking on Export Data to Clipboard. This will temporarily save the data so that it can be pasted into a software document of the user's choosing. Paste the data into an Excel file, then save the data. - Multiple spectra may be displayed in the window for comparison or for the purpose of making a figure. Drag the horizontal cursor (in the heat map, upper left) to a desired time point. Press Ctrl + S to select the spectrum and save it in the spectral window (lower left). Add as many time points as necessary to show the progression of the data (5-10 spectra), as seen in Figure 3.
NOTE: The number of spectra chosen to represent the data and the temporal arrangement of these spectra may depend heavily on the specific sample and experimental conditions. The recommendation above is a general guideline, but the specifics of the experiment should dictate which portions of the dataset are emphasized. - Export as data by right-clicking on the window containing the spectra. Click on the Export Data to Clipboard option. The data is temporarily saved. Paste this data into the desired software document (i.e., Excel) and Save.
- Multiple kinetic traces can be displayed in the kinetics window in the same manner as the spectra window. Drag the vertical cursor (in the heat map, upper left) to a desired wavelength. Press Ctrl + X to select the time trace and save it in the kinetics window (upper right). Add as many time points as desired. This will temporarily save the current kinetic trace in the window.
- Export as data by right-clicking on the window containing the kinetic traces. Click on the Export Data to Clipboard option. The data is temporarily saved. Paste this data into the desired software document (i.e., Excel) and Save.
- To export the heat map of the dataset, click on the File menu, then click on Export to CSV (Supplementary Figure 15). This will open a window, click on OK to save the csv file in the same directory as the data file open with the same name as the data file.
- Extracting data from single wavelength fitting for display
- Click on the Kinetics menu, then click on Fit Kinetic to open the window containing the fitted data.
- Right click on the fit window (i.e., the central tile in the single fit window). Click on Export Data to Clipboard. This will save it temporarily so that it can be pasted into a different software program.
NOTE: The residual plot below the fit data cannot be exported and will instead have to be recreated from the fit data. The fit exports both the raw data and the fit line, which can then be used to recreate the residual. The residual is created by subtracting the fit value from the data at each time point, and creating a plot similar to what is shown in the "Fit Kinetics" window. - Paste this data into the desired software document (i.e., Excel) and Save.
NOTE: Exporting to the clipboard will only include the raw data and the fit line data for each exponential used in the fit. The parameters of the fit, such as the lifetimes, amplitudes, and such, will not be included and will have to be exported by copying the values from the fitting software.
- Extracting data from global lifetime analysis for display and analysis
- Click on the Surface menu, then click on the Global Fit option to open the window containing the fitted data.
- The precision for the values of both the optical density and time delay/wavelength axes will need to be adjusted for the principal components (upper right tile) and DADS (lower left tile), respectively. Put the mouse over the principal component window until the setting box appears in the lower right.
- Quickly click on the x.xx button, mouse over "Precision," and click on 6 from the menu to set the number of decimal places to include.
- Put the mouse over the principal component window until the setting box appears in the lower right. Quickly click on the y.yy button, mouse over "Precision" and click on 6 from the menu to set the number of decimal places to include.
- Right click on the Principal Kinetic Traces window. Click on Export Data to Clipboard. This will save it temporarily so that it can be pasted into a different software program.
- Paste this data into the desired software document (i.e., Excel) and Save.
NOTE: The data will be saved as a series of columns containing first the Time Delays, then the Principal Kinetic Traces followed by the fit line. There will be one set for each principal component selected when preparing for global analysis. The DADS spectra have already been saved as part of the fitting procedure in step 2.4.7.
Preparation and analysis of a sample of 1,4-Bis[2-(5-phenyloxazolyl)]benzene, POPOP in ethanol was carried out following the procedure outlined above. Measurements were carried out using an ultrafast transient absorption spectrometer, as described in Figure 1, with fluid solutions in 2-mm cuvettes using an adjustable cuvette holder and a magnetic stirrer to ensure mixing. Samples were measured in ambient conditions with no additional controls for temperature or atmosphere. The optical window of 340 nm to 680 nm was generated using a calcium fluoride crystal. Two hundred fifty (250) time points were collected between −5 ps to ~5500 ps, and three scans were averaged to generate the final dataset, Figure 3. The POPOP data was prepared as described in the protocol. An example of sub-optimal chirp correction is shown in Supplementary Figure 16. Single-wavelength kinetic fitting was performed on POPOP, selecting 632 nm as the wavelength of interest. Additionally, global analysis was carried out on POPOP as described in the protocol.
Single-wavelength kinetic fitting of POPOP at 632 nm produced two lifetimes. These lifetimes were allowed to vary and no further adjustments were made. The final parameters obtained were as follows: t0 = −0.1176 ps, IRF = 0.436 ps, A1 = 0.0956, t1 = 1.614 ps, A2 = 0.0646, t2 = 522.2 ps (Figure 4). These results are in good agreement with global analysis carried out subsequently and reported emission lifetime values for POPOP (τ = 1.35 ns)17. An example of single-wavelength fitting with too few lifetime components is shown and discussed in Supplementary Figure 16.
Global analysis fitting on POPOP was performed after selecting 15 principal components (PCs) when performing SVD. Two lifetimes were selected after fitting, no parameters were fixed. The final parameters obtained from fitting were as follows: t0 = −0.1586 ps, tp (IRF) = 0.4408 ps, t1 = 1459 ps, t2 = 267.5 ps. The decay-associated difference spectra are displayed in Figure 5. Results were in good agreement with those from single kinetic fitting at 632 nm and lifetime values for POPOP17. Two examples of sub-optimal global analysis are shown and discussed in Supplementary Figure 16.
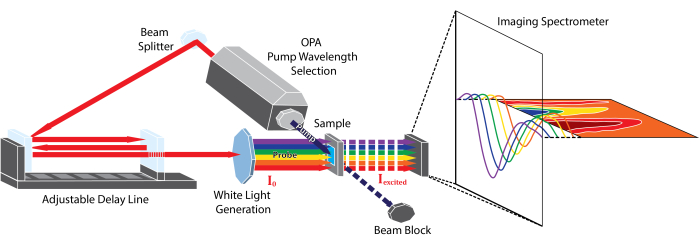
Figure 1: Schematic representation of the femtosecond broadband transient absorption instrument described in this protocol. Please click here to view a larger version of this figure.
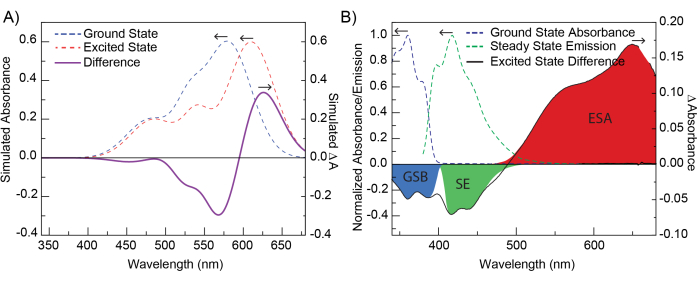
Figure 2: Simulation of TAS data and contributions to TAS signal. (A) Gaussian curves simulating a ground-state absorption spectrum (blue dotted line) and an excited-state spectrum (red dotted line, red-shifted from the ground-state spectrum). The difference spectra of these two spectra (purple, solid line) is the difference spectrum as viewed in TA. The differences between the ground- and excited-state spectra have been exaggerated for illustrative purposes. (B) A representative TA difference spectrum of POPOP at 1.04 ps after excitation at 330 nm. The dotted lines show ground-state absorbance and steady-state emission of POPOP. Highlighted regions show common TA features, ground-state bleach (GSB), stimulated emission (SE), and excited-state absorbance (ESA) observed within this data. Please click here to view a larger version of this figure.
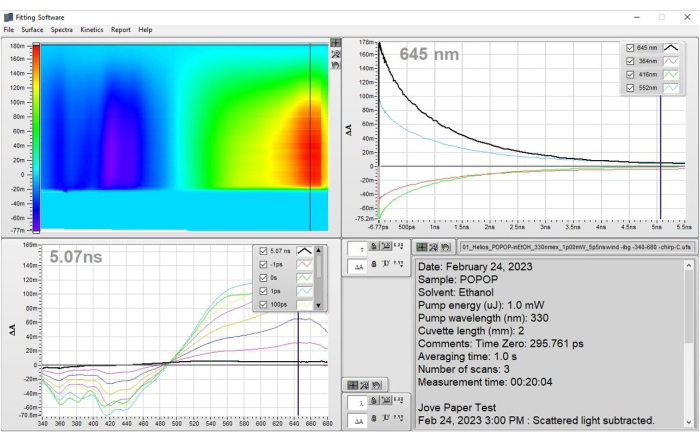
Figure 3: Prepared data for POPOP that result from the application of step 1 – Preparation of data. The data are shown as the corrected heat map and representative spectra. These results demonstrate how the data should look after the corrections have been applied and one is ready to apply a fit to the dataset. Please click here to view a larger version of this figure.
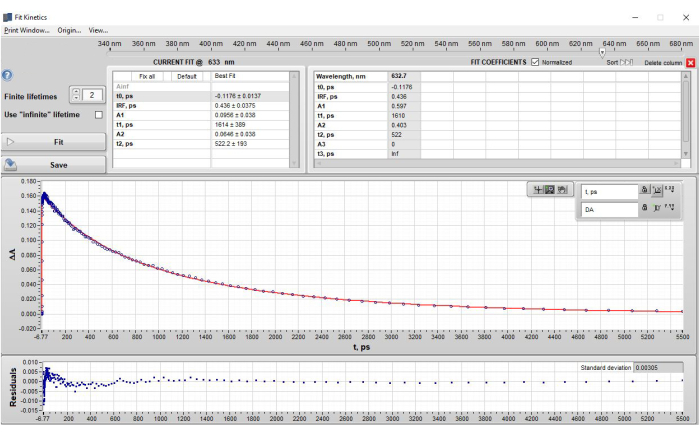
Figure 4: Results of single wavelength fitting of POPOP at 632 nm after application of step 2.3 – Single wavelength fitting. The figure shows (top section) the lifetimes obtained in a table format, (middle section) the data (blue dots) and the fit to the data (red fit line), and (bottom section) the residual plot. Note that under the "current fit" section, the amplitudes (A) are shown as the ΔA values from the data that represent the contribution of that specific lifetime component at t0. However, when the current fit is saved and appears in the "Fit Coefficients" Table, the default setting is to show the normalized amplitudes. This setting can be changed by unchecking the box next to the "Fit Coefficients" label "Normalized". Please click here to view a larger version of this figure.
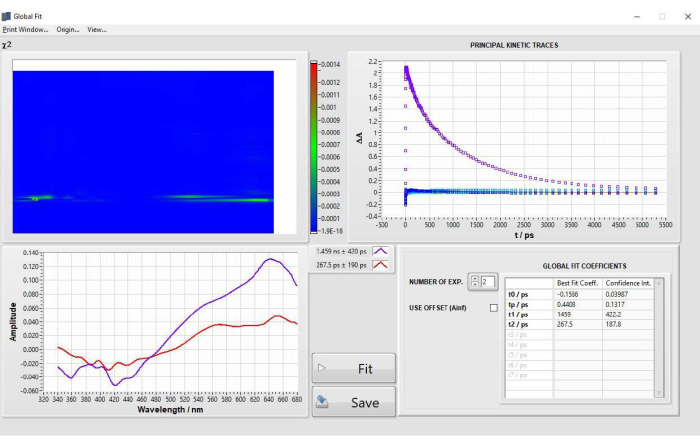
Figure 5: Results of global analysis fitting of POPOP obtained after step 2.4 fitting. Please click here to view a larger version of this figure.
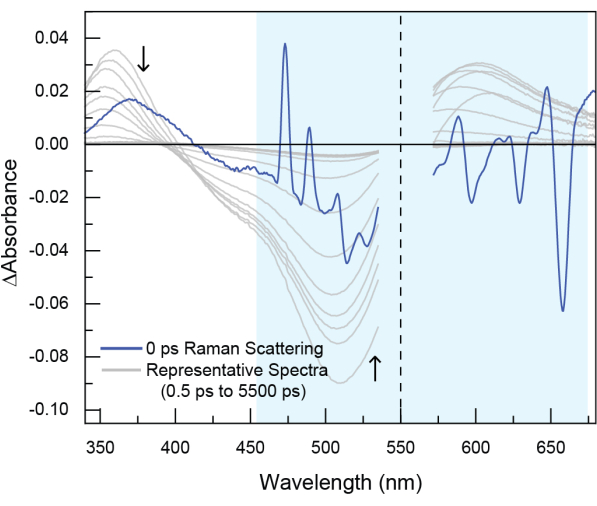
Figure 6: Example of stimulated Raman scattering observed in experimental TA data. In this dataset (not the POPOP data shown in the tutorial), the sample was excited at 550 nm (indicated by the dotted line). Raman scattering is seen around time zero and generally appears both to the blue (anti-Stokes Raman scattering) and to the red (Stokes Raman scattering) of pump excitation wavelength. Stimulated Raman scattering is short-lived, typically only around ~200 fs, because it occurs as the result of the probe beam interacting with the sample at the same time as the pump beam, thereby stimulating the Raman process. However, the feature cannot be avoided and should be removed with cropping. Please click here to view a larger version of this figure.
Supplementary Figure 1: Main menu when data is initially loaded. Generally, in this fitting software there will be option boxes that appear in the lower right corner of a window when the window is moused over. These boxes allow for alteration to the cursor interaction with the window, either moving selections, zooming, or panning the selected windows. There are also options for adjusting the precision of both the x-axis and y-axis scales for the window as well as switching the display from linear to logarithmic. The axes can also be locked or unlocked. While locked, an axis will remain at the zoom level or range of values specified; when unlocked, the range will switch to contain the full set of data. Other options allow for adjusting the display of numbers and the coloring of grids if present. Please click here to download this File.
Supplementary Figure 2: Surface menu to carry out scattered light subtraction. Please click here to download this File.
Supplementary Figure 3: Subtract scattered light screen – setting the spectra to average. Please click here to download this File.
Supplementary Figure 4: Setting cropping range. Please click here to download this File.
Supplementary Figure 5: Cropping dataset. Cropped data will be removed permanently. Please click here to download this File.
Supplementary Figure 6: Fit solvent response window and example of solvent response fit (in red line) to the data (blue dots). Please click here to download this File.
Supplementary Figure 7: Chirp correction window with "blank" sample data. Please click here to download this File.
Supplementary Figure 8: Chirp correction shown along with the dataset. Please click here to download this File.
Supplementary Figure 9: Single Kinetic Fit window. Additionally, a slider at the top allows the user to select the wavelength at which to perform the fit. Selecting an appropriate wavelength requires knowledge of the spectroscopy of the system to identify where processes of interest occur. Examples include charge-transfer products, triplet formation, or photoproduct formation, the spectral features of which are known to correlate with specific wavelengths. These specific wavelengths can be fit in order to obtain the lifetimes of those specific events. Additionally, a selection of wavelengths can be used in order to provide validation to a selected global fit model. Please click here to download this File.
Supplementary Figure 10: Setting Single Kinetic Fit parameters. Individual fit parameters can be fixed or altered by hand if desired to fine tune the fit and minimize the standard deviation of the residual. Note: Parameters can be easily adjusted by clicking on the value in the box and then adjusting using a slider or manually typing in a value. The displayed fitting will be adjusted in real-time as values are changed. When an acceptable fit is achieved, the fit can be exported by right-clicking on the fit screen, which allows exporting the data to the clipboard for pasting into a desired program or as a picture for rapid viewing. Please click here to download this File.
Supplementary Figure 11: Constraining and fixing values of Single Kinetic Fit parameters can be done if one or more parameters are well-known. Please click here to download this File.
Supplementary Figure 12: Single Value Decomposition window with Principal Components showing how the trace and components will look when enough principal components are added. Please click here to download this File.
Supplementary Figure 13: Global Fit window before global analysis fit is generated. Please click here to download this File.
Supplementary Figure 14: Global Fit window showing the results of the global analysis fitting. Please click here to download this File.
Supplementary Figure 15: File Menu for saving and exporting files. Please click here to download this File.
Supplementary Figure 16: Examples of sub-optimal chirp correction and fitting. (A) demonstrates a poor single kinetic fit. The type of structured residual shown in panel A typically indicates that an additional lifetime is required to fit the data. Notice that the residual dips below zero at shorter times and is raised above the zero line at longer lifetimes. (B) demonstrates a correct chirp correction using the preview button, in which features are straightened and no curvature is present in the data. (C) demonstrates an incorrect chirp correction in which there is noticeable curvature present in the blue portion of the spectrum, indicating that the chirp function is overcorrecting in this region. (D) displays a poor global lifetime analysis fit in which overfitting (including too many parameters) has resulted in "symmetrical" DADS (that look like mirror images of each other across the x-axis) of similar lifetime ranges that essentially cancel each other out. When these features are observed, the fit should not be used. (E) displays a poor global fit in which too many parameters have resulted in a very short lifetime with a very large amplitude. The issue present in (E) may also appear if artifacts around time zero are not properly corrected and the fitting becomes focused on minimizing the residual by overemphasizing the very short lifetime (one that is not physically meaningful). Please click here to download this File.
Supplementary Figure 17: Example of TAS data with Raman scattering. Scattering is present around time zero, and coincides with the pump excitation wavelength. Scattering consists of a series of sharp peaks with a very intense positive peak blue of the pump excitation and a negative peak red of the pump excitation. This feature cannot be prevented reasonably and should be cropped out of the data to avoid interfering with the fitting results. Please click here to download this File.
Supplementary File 1: File that contains the dataset for this tutorial (POPOP data_POPOP-inEtOH.ufs). Please click here to download this File.
Supplementary File 2: File that contains blank dataset for this tutorial (POPOP data_BLANK.ufs). Please click here to download this File.
Supplementary File 3: Supporting Information files containing additional comments on fitting solvent response, correcting for scatting, and subtracting surfaces. Please click here to download this File.
General considerations for data preparation
Fitting TA data may appear at first glimpse to be relatively straightforward, and it might be expected that one clear correct "answer" should result for a given dataset. However, as highlighted in the protocol, there are many factors in the data acquisition, data preparation, and data analysis to carefully consider that can lead to uncertainty over which model or set of fitting parameters best describes the data. The goal of data preparation and fitting is to reduce as many of these extraneous factors as possible, while preserving the data for analysis. The task at hand might seem daunting to a beginner, as there is much to consider. To build intuition about the fitting process, the beginner is encouraged to try preparing the same data multiple times from scratch in slightly different ways to check how dramatically the data preparation steps impact the best fit. Additionally, two different researchers can prepare and fit the same data and compare results. This process may be time-consuming the first several times, however, doing so will allow the beginner to develop intuition on how to consistently prepare data for future samples. Like any skill, this data preparation and fitting will take time to develop, and the beginner is encouraged to be patient and disciplined when experimenting with and learning the process. The dataset used in this study is provided to give the beginner the chance to fit directly alongside the tutorial, and to directly compare results with those produced in the tutorial.
The data may contain background features that are present at all time delays (Supplementary Figure 2 and Supplementary Figure 3) such as scattering of the pump beam, and spontaneous emission of the sample. These unwanted features must be removed in order to isolate the transient absorption signal from the species of interest11. Removal of such features is done by choosing, averaging, and removing the contribution of a number of negative-time difference spectra. When selecting background spectra, it is important to ensure no features that might be part of the process of interest are included for removal. Background features arising from the solvent, such as absorption from impurities or the solvent itself, may also be observed in TA data. When the solvent produces a signal, a "blank" dataset containing only the solvent run under the exact same experimental conditions as the sample will need to be subtracted from the sample dataset. Details on this procedure are included in Supplementary File 3.
The chirp correction is another factor to consider carefully. Chirp occurs as the probe pulse travels to the sample and broadens due to imperfections in steering mirrors or by passing through dispersive optics such as lenses or filters. The end result is that lower energy photons in the probe pulse (i.e., the red side of the probe spectrum) arrive at the sample before higher energy photons (i.e., the blue side of the probe spectrum). This results in the "time zero" of the TA spectra being smeared out over several femtoseconds or picoseconds18, which manifests as a distinct curve in the raw dataset starting in the blue wavelengths and then flattening out as it approaches the red (Supplementary Figure 7). Chirp is most noticeable on shorter time scales such as those accessed by ultrafast TA. This wavelength-dependent time zero can be corrected as described in the protocol, but the application of this process can be tricky and subjective. Having a "blank" sample or measurement of the solvent Kerr response can minimize the subjective nature of hand-picking points for the chirp correction needed to generate the polynomial fit used to adjust and correct the chirp. The goal of the chirp correction is to remove the distinct "curve" of the time zero. It may take multiple attempts at fitting the chirp to obtain the best chirp-corrected data. The data can be fit several times with different chirp corrections applied in order to gain an understanding of the impact the chirp correction has on the values of the short TA lifetimes.
Artifacts that appear at "time zero"
Several artifacts can be observed close to "time zero" in TA data, including Rayleigh scattering, stimulated Raman scattering and cross-phase modulation. Rayleigh scattering of the pump beam is elastic scattering that results without a change in energy. This feature will appear at the same wavelength as the pump pulse. Stimulated Raman scattering may accompany the pump scattering signal19. Raman scattering, which results from inelastic scattering of a pump photon, produces peaks at both higher (anti-Stokes) and lower (Stokes) energy than the incident pump energy. In TA data, stimulated Raman scattering is observed due to the simultaneous irradiation of the sample with the pump and probe beams. When the probe beam interacts with the sample at the same time as the pump beam, it stimulates the Raman process. Therefore, the stimulated Raman scattering occurs around time zero and results in additional peaks in spectra within the first few hundred femtoseconds (Figure 6, observed in the darker blue spectrum in the highlighted region and Supplementary Figure 17). Cross-phase modulation originates from modulation of the solvent refractive index from interaction with the intense electric field of a pulse.
Stimulated Raman scattering may be distinguished from cross-phase modulation because the Raman peaks appear at specific frequencies that correspond to vibrational modes of the solvent. Because it is a Raman process, both Stokes and anti-Stokes lines on either side of the excitation can be observed. Chlorinated solvents like methylene chloride show very prominent Raman bands due to the large polarizability of chlorine. The spectral signatures of cross-phase modulation are unique to a solvent but are not as easily predicted as Raman scattering features.
Depending on the kinetics of the sample being measured, Rayleigh scattering, Raman scattering, and cross-phase modulation may overlap with early features of the TA data and can be challenging to remove from the data. In principle, these features can be seen in a neat solvent measurement and subtracted from the data, data analysis programs may have fitting functions to account for these features, but in practice, this can be difficult. When it is too difficult to subtract these artifacts without compromising the sample data, it may be better to crop out the compromised spectra around time zero to eliminate the artifacts. Doing so will have the unfortunate side effect of removing the first approximately 300 fs of data but will make fitting more reliable later on. Over the course of analyzing multiple datasets of the same and different samples, the beginner will gain intuition in achieving this balance of subtracting the background surface versus cropping out the initial 100-200 fs data.
General cropping may be necessary for portions of the spectra that contain low signal-to-noise. Instability in the probe beam at certain regions, low intensity of probe light, sample concentrations that are too high (thereby blocking much of the incident probe), low pump intensity, and the absorption cross-section of the sample are typical culprits of low signal-to-noise that can make fitting data challenging. In these cases, cropping the dataset on either side of the optical window in order to achieve a desired level of signal-to-noise can help the fitting process.
A dataset is ready for analysis once it has been sufficiently cropped to remove poor sections of the dataset, had the chirp corrected, and had background spectra averaged and subtracted. This procedure should result in data that contains only those portions most relevant to the photophysics and photochemistry of interest. Indeed, it is clear that there is some degree of subjectivity to this process. The goal in data preparation is to strike a balance of removing artifacts so they do not perturb the fitting, but not to remove so much that it compromises the integrity of the dataset, thereby hindering its interpretation. Finding this balance takes time and experience to build up the intuition for what is an artifact and what is data. Fitting (and re-fitting) the same set of data on multiple different days, or having two researchers fit the same data, can be a way to minimize human error and the subjectivity of data preparation and analysis.
General considerations for fitting and interpretation
After the raw TA spectra have been processed, they must be interpreted and modeled to extract information about the species and the dynamics present in the system of interest. This process can be described as a three-step procedure that includes initial spectral interpretation, quantitative modeling/fitting, and assignment of the spectral interpretation to the model/fitting.
Initial Spectral Interpretation: In the spectral interpretation step, the goal is to assign features present in the TA spectra to electronic states accessed in the photophysical or photochemical evolution of the system. To begin, various states should be identified. In this work, states refer to unique electronic states that are part of the photophysical or photochemical evolution of the system. A state, represented, for example, by one specific potential energy curve (PEC), possesses a set of characteristic peaks representing its absorption spectrum. A change that occurs within a single state is called a process. A photophysical process may appear in TA spectra as a peak shift or a change in the width of the spectrum. The key aspect of a process is that the population of the state stays the same (i.e., the process occurs within a given PEC); it is the distribution of energy within the state that changes. A change in the population of a state will be referred to as a transition. During a transition, the system evolves to another PEC (i.e., electronic state). Transitions may include internal conversion (IC), intersystem crossing (ISC), charge transfer, energy transfer, formation of new products, or return to the ground state. Guidelines for assigning states, processes, and transitions are discussed in the following paragraphs.
Assigning states
The first step in this process involves assigning spectral features to specific chemical species or states. The S1 state in TA should show a lifetime that matches the fluorescence lifetime taken using time-resolved emission spectroscopy. A triplet state can be verified if its lifetime is quenched by oxygen. If a radical anion or cation is suspected in the photophysical evolution, spectroelectrochemistry or chemical oxidation/reduction can be performed to generate the radical species, and an absorption spectrum of that species can be obtained and compared to the TA bandshape. Electron spin resonance (ESR) spectroscopy can be performed to verify the presence of free radicals. An excellent tutorial talk hosted by the ACS Division of Inorganic Chemistry gives an overview of TA and such considerations in assigning features20. After bands have been assigned to species, the next step in interpreting TA spectra is to qualitatively describe the dynamical processes occurring in the system. This step is vital as it gives the researcher an idea as to what models will be appropriate for describing their system and will give them a baseline to compare the fit parameters to.
Changes within a state
Vibrational cooling, geometric rearrangement, or solvation are extremely rapid processes (sub-ps to 10's ps) that can be observed with TA. Vibrational cooling is observed as rapid blue shifting of the TA spectrum on a several picosecond timescale21,22,23. Geometric rearrangement can occur on the 10's ps timescale. Solvation dynamics are observed as a redshift and narrowing of the spectrum over several picoseconds in conventional dipolar liquids, but high-viscosity solvents such as glycerol, polyethylene glycol (PEG), ionic liquids, and deep eutectic solvents can exhibit solvation dynamics occurring over the course of multiple nanoseconds24,25,26.
Changes in a state population
Reactions are characterized by a change in the intensity of a band, where a decrease in intensity is associated with a decrease in concentration of its chemical species and vice versa for an increase. In some cases, both the reactant and product species are visible in the spectra, whereas in others, the product states are too short-lived or too far red-shifted to be observed. Often state-to-state transitions can be observed by the presence of an isosbestic point in the spectra.
Quantitative modeling/Fitting: A model must then be fit to the data in order to extract quantitative information about the dynamics of the system. As previously described in the introduction, there is a vast array of models to use. This protocol focuses on two of the most common methods: single-wavelength fitting and global analysis. The single-wavelength method involves fitting individual wavelength traces from the spectra to some functional form, typically a sum of exponentials:
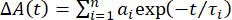 (2)
(2)
where ΔA(t) is the TA signal at a chosen wavelength, n is the number of exponential components, and ai is the amplitude of exponential component, i, with time constant τi. Several components can be added until the fit reproduces the experimental data. The goal of any fitting process is to model the data using enough lifetimes to reproduce the data well, but not overfit the data by including too many components. Hence, weighted goodness-of-fit parameters such as , are used to help determine when the data are fit to within experimental uncertainties5.
After the decay is fitted satisfactorily, the parameters of the model can be used to characterize the dynamics of the system. The resulting time constants can then be extracted and interpreted. Unfortunately, the large number of overlapping features in TA spectra mean that a single wavelength in the spectrum may contain dynamics corresponding to different species whose spectral signatures overlap, meaning that the time constants extracted from a single wavelength fit may represent a composite of multiple coinciding processes. Additionally, any changes in bandshape and position will also influence the amplitudes and time constants extracted from single-wavelength fitting. These issues can be circumvented in some cases by a fitting method called 'bandshape analysis,' where one determines or assumes a functional form for the TA bands of each absorbing species in the system. These shapes are then weighted by time-dependent amplitudes and summed together in order to reproduce the observed spectrum. This procedure is commonly used in the analysis of time-resolved fluorescence spectra, but the more complicated shapes and overlapping components of TA bands make this method tenable in only a few simple cases, as detailed elsewhere10.
Another drawback of single-wavelength fitting is that it does not intrinsically take advantage of the broad spectral range afforded by modern TA experiments. One could, in principle, methodically fit each individual wavelength of the spectra, but such analysis is cumbersome, time-consuming, and computationally expensive. To combat this challenge, a method called 'global analysis' can be used to simultaneously fit an entire set of TA spectra to a set of shared dynamical parameters4. Global analysis, and a closely related method called target analysis, are successful and widely used methods, but they also come with their own unique set of drawbacks and limitations. As with any model, it is imperative to understand the assumptions that are used to create it as well as the limitations that they present.
In global analysis, TA spectra are represented by a m by n matrix, where m represents the number of wavelengths measured in each spectrum and n represents the number of time points collected. This matrix is then assumed to be decomposable into the product of two other matrices:
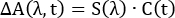 (3)
(3)
where C(t) is an n by k matrix and S(λ) is an m by k matrix. The value k represents the number of distinct spectral components used to reproduce the spectra. Each of these components represents an absorbing species with a unique spectral signature and dynamics. The S(λ) matrix represents the TA spectra of the k components and C(t) their time-dependent concentrations. In the simplest and most common implementation of global analysis, each component is assumed to have single-exponential kinetics (i = 1 in Equation 2, with each component assigned its own time constant). In summary, the full TA spectrum can be represented by the sum of k spectra components, each with its own characteristic absorption spectrum and single exponential decay.
When the TA spectra are fit, the user guesses how many components (i.e., a value for k) are needed and makes a guess at the time constant associated with a single-exponential decay of those species. The fitter then generates Cguess(t) and solves Equation 3 for Sfit(t). Next, Sfit(λ) and Cguess(t) are multiplied as in Equation 3 to create the fitted spectra, ΔA(λ,t)fit. Finally, the residuals, ΔA(λ,t)exp − A(λ,t)fit, are minimized and the optimal Sfit(λ) and time constants returned. The relative simplicity of global analysis, representing an entire set of spectra using a handful of time constants and fixed spectral components, makes it an attractive (and successful) method for untangling the complicated bandshapes and dynamics encountered in TA spectroscopy. However, care must be taken to ensure that global analysis is an appropriate model for the system at hand.
A key assumption in global analysis, illustrated in Equation 3, is the complete separability of the wavelength and time portions of the dynamics, a property called 'bilinearity'. This assumption requires that the component bandshapes are time-independent (i.e., they have a fixed spectral shape that does not vary or shift with time). The only thing that changes during the experiment is the relative populations of each component, represented by C(t). On long time scales, ~1 ns or so, this assumption typically holds and global analysis can be used without much concern. On the other hand, excited-state processes such as vibrational cooling and solvation dynamics, prominent on the ultrafast timescales accessible by femtosecond TA, result in time-dependent changes in the spectral signature of a species and a breakdown of bilinearity. This does not mean that global analysis cannot reproduce a dataset, in fact, it can always produce a satisfactory fit provided a sufficient number of components are used. The problem then lies in interpreting the component spectra and assigning the time constants to particular excited-state processes, as the components may no longer correspond to distinct absorbing species. Therefore, care must always be taken when applying global analysis to situations when bilinearity cannot be assumed.
Assigning the spectral interpretation to the model/fitting: Once a fit is obtained, the spectral interpretation must be mapped onto the lifetimes obtained in the fit. The lifetimes from the fit are assigned to both processes and reactions that were identified in the initial interpretation of the spectra. However, the initial assessment from the spectra and the number of fitted lifetimes obtained by the model might not immediately map onto each other. In this (common!) situation, the fitter needs to go back and assess the initial interpretation. Perhaps there was a vibrational cooling or other process that was missed in the initial assessment, but was identified in the modeling and fitting process. Or, perhaps two different sets of fit parameters could reproduce the data well and the initial interpretation can guide which set of fit parameters are chosen. In this final step, the fitter must go back and forth between the interpretation and fitting to find a description that leads to a plausible photophysical assignment of the species and dynamics of the system. Other fitting programs that include sequential fitting models, such as target analysis, can also be explored to complement the fits yielded by global analysis and the fitting software presented in this article4.
In summary, this protocol discusses the preparation and fitting of transient absorption data. Its aim is to highlight challenges associated with the process and to comment on ways to avoid or mitigate these challenges in a practical manner. Fitting TA data, like fitting most data encountered in technical fields, can be tricky and, at times, subjective. Therefore, being aware of the process and limitations of the data, the data preparation, and the mathematical tools used to model and assign meaning to the data are critical. Scientists must approach data and modeling with a critical eye.
One can attempt to mitigate the subjectivity of their fits. For example, the data can be prepared and fit from different starting points and on different days to ensure that the same fit is produced. Data taken on different days with different sample preparation can be compared. Multiple researchers can fit the same data and compare their results. Over time, researchers can build an intuition about the data they obtain (based on the specifics of their experimental setup and experimental parameters) that will allow them to be more confident in their fits.
There is a great deal to learn about TA data fitting and the details of the models discussed in this article. Several excellent review articles are enthusiastically recommended that delve deeply into this topic4,10,27. This protocol is meant to be a beginner's entryway into the analysis and fitting process that spurs interest in more deeply understanding the process.
