For the migration assay, it is crucial to thoroughly examine the images captured at the t = 0 h time point to ensure the presence of a fully formed cell monolayer is accurately detected by the system (Figure 1B). Additionally, the clarity and straightness of the scratch border should be confirmed (Figure 1B). The cell-free area ought to be largely free of debris. At the end of the assay, a group stimulated with, for example, 25 ng/mL VEGF as a positive control should show successful migration in the original scratched area (Figure 1B). An instance of technical issues might manifest as patchy cell growth, double scratching, or waviness in the scratch borders (Figure 1C). Furthermore, it is important to establish a substantial dynamic range between the negative control (EBM-stimulated samples) and the positive control (VEGF samples) (Figure 1D). In our experimental setup, we consider a 25% increase in RWD after 12 h as an optimal range (Figure 1D). In a technical problematic assay, positive and negative controls do not separate correctly, indicating a low dynamic range (Figure 1E).
Immunocytochemical staining of HUVECs on coverslips, visualized under a fluorescence microscope to validate successful staining (Figure 1F). It is important to survey a majority of the cells and identify a characteristic region of the slide. Staining specificity is assessed using a control slide treated only with a secondary antibody to exclude nonspecific binding (Figure 1F). Specific staining signals should be critically examined according to the known subcellular localization of the target protein. We use subcellular localization patterns along with the absence of signal in the control sample as indices of a successful immunostaining protocol.
Similarly, certain aspects must be considered for high-quality spheroid sprouting assays. The quality should, in this case, be assessed at the end of the assay at, for example, t = 24 h. The collagen matrix observed under the microscope should exhibit homogeneity and should not appear friable or rough (Figure 2B). Spheroids stimulated with just basal medium as a negative control and 25 ng/mL VEGF as a positive control should be clearly distinguishable (Figure 2B). Furthermore, spheroids should float in the gel and not drop to the ground, where they disperse, indicating low quality (Figure 2C). This issue often arises from inadequate cooling of the collagen during titration of the mixture or from over- or under-titration of the pH (Figure 2C). A minimum of 10 spheroids per well should be imageable for each well. The negative control (EBM-stimulated samples) should demonstrate a moderate baseline sprouting rate, while VEGF, serving as the positive control, ideally doubles or, preferably, triples the RSL compared to the controls (Figure 2D, left graph). The number of sprouts should also show a similar dynamic range (Figure 2D, right graph). A technical suboptimal assay will not show this dynamic range between controls and could, therefore, lead to a false interpretation of data (Figure 2E).
Analog to 2D stainings, examination of the slides under a fluorescence microscope and critical assessment of the stained structures are of great importance for successful staining (Figure 2F). If the cellular localization of the structures and the signal intensity in comparison to the control is comprehensible, it can be assumed that the staining was successful.
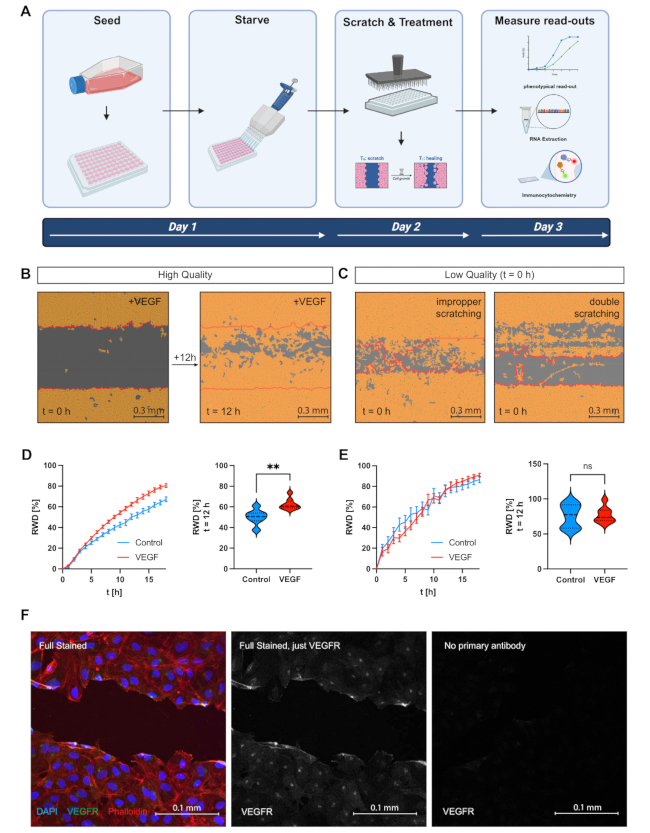
Figure 1: The live-cell imaging scratch wound migration assay. (A) Scheme of the scratch wound migration assay. (B) Images showcasing a high-quality technical replicate at t = 0 h and t = 12 h. Cells have been stimulated with 25 ng/mL VEGF to serve as a positive control. (C) Examples of two low-quality technical replicates at t = 0 h due to improper scratching or double scratching. (D) Results of a successful scratch wound migration assay. The correct separation of positive control stimulated with 25 ng/mL VEGF and negative control just treated with basal medium was further visualized by a violin plot at t = 12 h. Data include 7-8 technical replicates per group. (E) Exemplary results highlighting a failed Scratch Wound Migration Assay. Suboptimal separation of positive control stimulated with 25 ng/mL VEGF and negative control just treated with basal medium was visualized by a violin plot at t = 12 h. Data include 7-8 technical replicates per group. (F) Exemplary images of HUVECs seeded on coverslips and stained for DAPI, phalloidin, and VEGF-R2. Please click here to view a larger version of this figure.
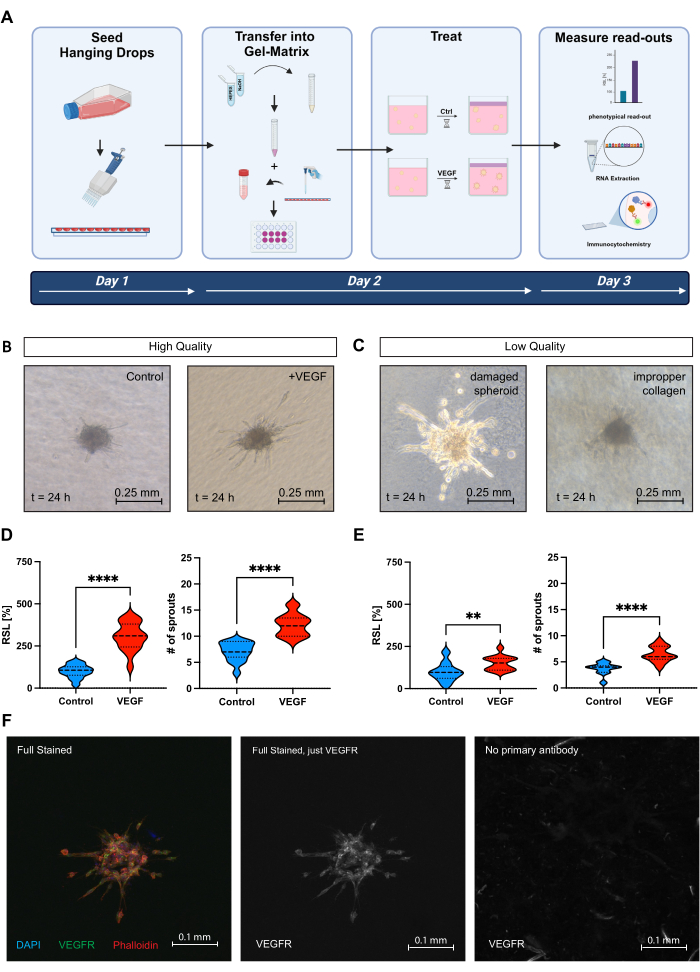
Figure 2: The spheroid sprouting assay. (A) Scheme of the spheroid sprouting assay. (B) Images showcasing the negative control (basal medium stimulated spheroids) and positive control (VEGF 25 ng/mL stimulated spheroids) of an ideal technical assay at the end at t = 24 h. (C) Examples of two low-quality spheroids at t = 24 h. Left images visualize a damaged spheroid that dropped to the bottom of the plate, which results in a loosened spheroid body and an abnormally enhanced sprouting rate. The right image visualizes collagen with noticeable color patches, creating a highly heterogeneous environment that significantly compromises the assay's quality. (D) Analysis of the RSL (left graph) and number of sprouts (right graph) of a successful assay with a good dynamic range between negative and positive control. Data is visualized using a violin plot and includes 10-25 spheroids per group. (E) Analysis of the RSL (left graph) and number of sprouts (right graph) of a technical problematic assay with a suboptimal dynamic range between negative and positive control. Data is visualized using a violin plot and includes 10-25 spheroids per group. (F) Exemplary images of HUVECs from the spheroid sprouting assay stained for DAPI, phalloidin, and VEGF-R2.c
Supplementary File 1: Instructions for preparing the methocel stock solution. Please click here to download this File.
Supplementary File 2: SpheroidCount.ijm plugin. Please click here to download this File.
