Here we performed ex vivo live imaging of single cell divisions within the E8.5 mouse neuroepithelium. To label individual cells, we induced Cre recombinase in a subset of cells containing a Cre reporter line that expressed dsRed upon recombination 5,6(Figure 3A). Thus, 48 hr later we were able to observe single cell divisions during ex vivo imaging (Figures 4A-D). Concomitantly, we monitored when the labeled cells became Shh-responsive by including a Shh transcriptional reporter modified BAC transgenic Olig2-eGFP line (Figures 3C-D; Figures 4G-N) 1-3. To observe cilia formation we generated Sstr3-GFP lentivirus to infect embryos in culture (Figure 3B; Figures 4A-D) 4. Immunofluorescence was carried out to confirm in vivo results (Figures 4E-F).
The time-lapse confocal imaging observation of the dsRedCre reporter embryos expressing Olig2-eGFP or infected with Sstr3-GFP lentivirus during in vitro culture are showed in Figure 5 10. In order to be sure that the Sstr3-GFP expression we were using to visualize cilia was not interfering with any underlying biological process, we corroborated our live imaging data with fixed wild-type embryo sections. Because we found the same percentage of asynchrony in cilia formation and Shh response, it indicates the SSTR3-GFP virus did not impact these processes (Figure 5A).
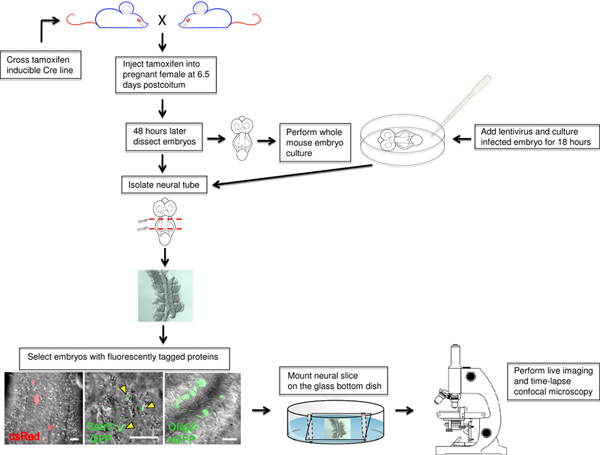
Figure 1. Flow chart of the ex vivo live imaging procedure using mouse neuroepithelium. The final mouse cross and tamoxifen treatment are depicted followed by the whole mouse embryo culture method. Embryos expressing fluorescently tagged proteins are selected and prepared for live imaging. Click here to view larger figure.
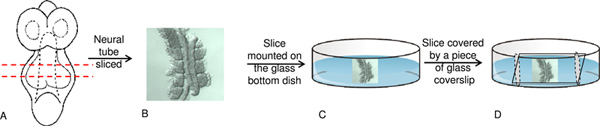
Figure 2. Neural tube slice preparation for live imaging. A) E8.5 unturned embryo with first appearance of somite pairs. B) Neural tube dissected in pre-warmed medium using a micro-knife. C) Neural tube slice is mounted ventral side down on the poly-L-lysine coated glass bottom dish. D) Small amount of a mixture made from petroleum jelly and wax applied around the neural tube and gently covered by a narrow piece of glass coverslip. Click here to view larger figure.
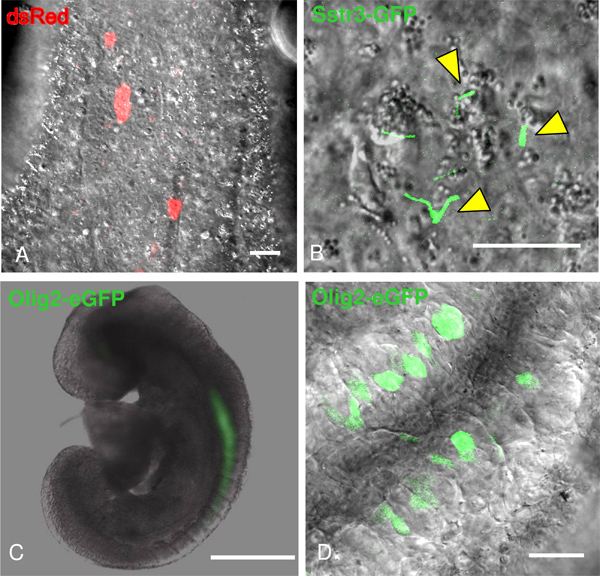
Figure 3. Visualizing fluorescently tagged proteins in live cells of the neural tube with confocal microscopy. A) DsRed labels individual cells. B) SSTR3-GFP lentivirus expressed in primary cilia as they form (yellow arrowheads). C-D) E8.5 embryo carrying Olig2-GFP shows GFP expression within neuroepithelium. Bar is 30 μm (A, D), 15 μm (B) and 1.5 μm (C).
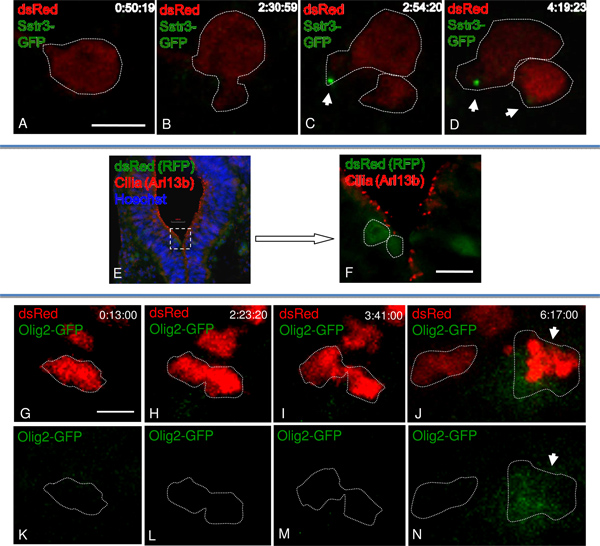
Figure 4. Monitoring cilia formation and Shh signaling in dividing cells of the developing neural tube. A-D) Single dsRed positive cell undergoing division shows a cilium forms in one daughter cell prior to the other cell (C, white arrow). The movie was imaged at a rate of one frame every 10 min. E-F) Immunofluorescence using antibodies against RFP in green (for dsRed lineage tracing) and Arl13b in red (for cilia). Enlargement of boxed area (E), without Hoechst (F). G-N) The dsRed positive cell undergoes division (I-M). The Olig2-GFP is expressed only in one daughter (J, N). The recording was imaged at a rate of one frame every 10 min. Bar is 5 μm (A-D), 50 μm (F), 25 μm (G-N).
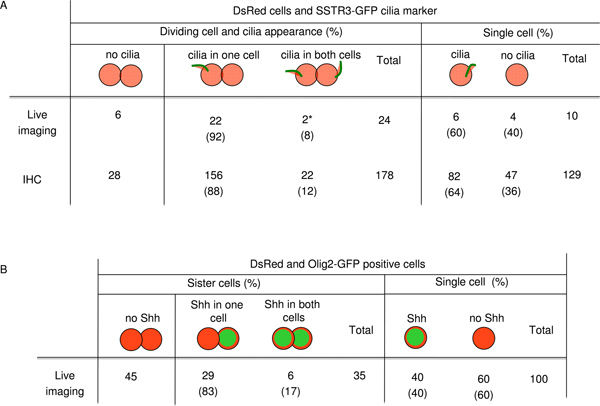
Figure 5. Counting of dsRed positive cell with cilia and Shh appearance. A) Number of dsRed cells undergoing division and cilia localization. Symbol * means by live imaging cilia formation was synchronous. B) Number of dsRed and Olig2-GFP positive cells undergoing division and Shh signaling.
