Measuring the affinity of non-phosphorylated STAT3 protein binding to oligonucleotides.
HEK293 cells expressing STAT3-GFP were used as a source of fluorescently labeled STAT3 for DNA binding assay. Cell lysates were prepared using RIPA buffer (20×106cells/ml). For binding studies, the lysates were diluted 150x with MST DNA-binding buffer to provide the optimal level of the fluorescent protein in the binding reaction (about 20 nM). Non-transfected HEK293 cells have been used to evaluate background fluorescence, which turned out to be non-detectable even in non-diluted lysate. However, background fluorescence can be more significant in other expression systems and thus has to be monitored. Titration series consisting of 11 binding mixtures and lysate sample without the ligand have been prepared. Each sample contained 15 μl of diluted cell lysate and 15 μl of oligonucleotides solutions of varying concentrations. Final buffer composition included 25 mM HEPES, pH 7.2; 50 mM NaCl; 2.5 mM MgCl2; and 0.025% NP-40. The measurements were taken in standard treated capillaries on Monolith NT.115 instrument using 50% IR-laser power and LED excitation source with λ = 470 nm at ambient temperature. Binding of highly charged oligonucleotides resulted in significant changes in STAT3 mobility in temperature gradient (Figure 2). In Figure 3, thermophoretic signal is plotted as a function of oligonucleotide concentration. Each data point represents the mean of three measurements. NanoTemper Analysis 1.2.20 software was used to fit the data and to determine the apparent KD values. The apparent dissociation constants were 37.9±1.0 μM and 23.3±0.6 μM for GAS and S+100, respectively (Figure 4). Substitution of A-to-G resulted in dramatic decrease in affinity of S+100 mut#1 (Figure 4), while mutant S+100 mut#2 showed no detectable binding thus confirming sequence-selective binding of STAT3 to S+100. Surprisingly, S+100 sequences displayed slightly tighter binding than GAS in three measurement repetitions.
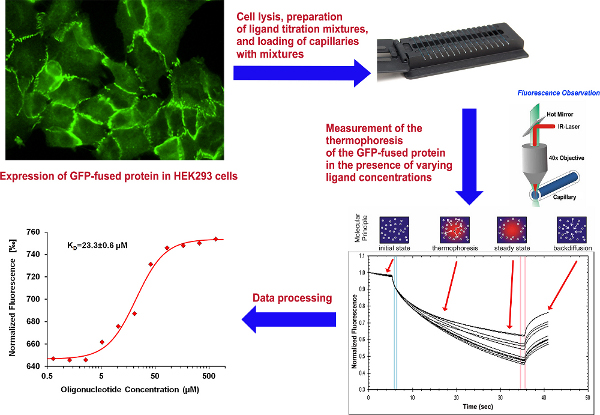
Figure 1. Overall scheme of the experiment. Click here to view larger figure.
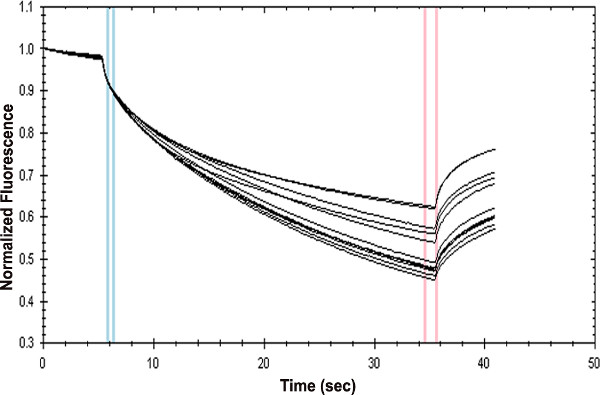
Figure 2. Unprocessed thermophoresis data generated for the interaction of GFP-STAT3 with AT-rich oligonucleotide. Diluted cell lysate containing 20 nM GFP-STAT3 was mixed with increasing amounts of double-stranded oligonucleotide (5′-AAAACAAAACGAAACAAACAAACTA) yielding the specified concentrations of the ligand. The data were collected at 50% laser power and 100% LED.
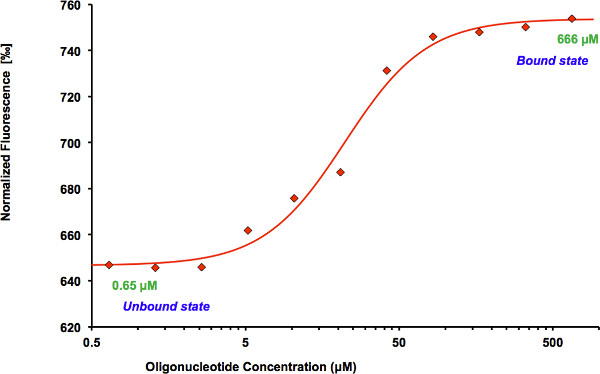
Figure 3. Binding curve generated by the NanoTemper Analysis 1.2.231 software. Normalized fluorescence (hot fluorescence/initial fluorescence) is plotted as a function of oligonucleotide concentration. The protein shows an increase in fluorescence in the bound compared to the unbound state. The data are fitted using the Hill equation method incorporated into the NanoTemper Analysis software.
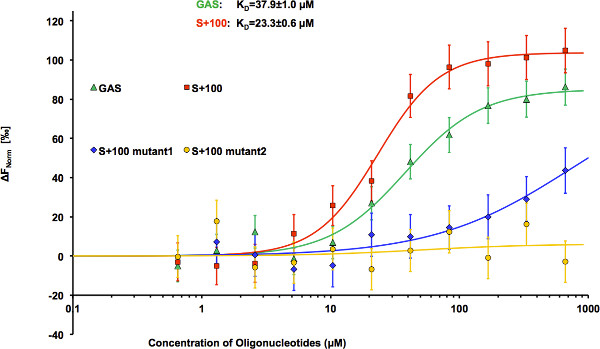
Figure 4. STAT3 binding to oligonucleotides with different sequences. Microscale thermophoresis binding measurements of STAT3-GFP to GAS (KD = 37.9±1.0 μM), S+100 (KD = 23.3±0.6 μM), S+100 mutant 1 (KD = 740±21 μM), and S+100 mutant 2 (no binding). The STAT3-GFP concentration was kept constant at about 20 nM, and the concentration of oligonucleotides varied from 666 to 0.650 μM. The difference in normalized fluorescence [‰] is plotted as a function of oligonucleotide concentration, and curves are fitted using the Hill method of the NanoTemper Analysis software. Error bars represent standard error of 3 measurements.
