Grafting
EpiSCs that ubiquitously express EGFP (r04-GFP, derived from E6.5 epiblast, and C2, derived in vitro from mESCs)4 were manually scraped from the culture dish and grafted into different sites of E7.5 embryos (Figure 2A). The embryos were cultured ex vivo and analysed after 24 hr. The distribution of donor cells was assessed by fluorescence microscopy. If donor cells incorporated, they proliferated and their derivatives dispersed within the host embryos (Figure 2B). It has been observed that grafts containing 10-16 cells incorporated efficiently in the host embryos (Figure 2A, and 2B), however, grafting more cells does not result in better chimaerism. Instead, grafted cells produced unincorporated clumps (Figure 2C and 2D).
Electroporation
To assess the efficiency of our electroporation system, we delivered GFP-expressing plasmids (pCAG-GFP and pCAG-Cre:GFP) to specific sites in the embryo. Consistent with a previous study11, GFP+ cells were detected in embryos 1-2 hr after electroporation (Figure 2E and 2G). When distal epiblast cells at the late primitive streak stage embryo were electroporated, labelled cells contributed to the neural ectoderm after 24 hr in culture (Figure 2E and 2F). This result corresponds well to known fate maps of epiblast cells in gastrulation stage embryos17. Similarly, when the GFP expression plasmid was electroporated in the primitive streak at E8.5 (2-5 somites), GFP+ cells contributed to the paraxial mesoderm (Figure 2G and 2H), consistent with known fate maps of the late primitive streak18. Moreover, we observed contribution to all three germ layers from electroporated cells (Figure 2I-K), suggesting that the electroporation procedure does not compromise cell behaviour in vivo. However, we also noticed that whilst epiblast (E7.5) or primitive streak cells (E8.5) were targeted, some endoderm cells were also electroporated (Figure 3C and Table 1).
One of the major advantages of using a capillary electrode is that the number of electroporated cells can be controlled, simply by changing the diameter of its opening. To determine the number of electroporated cells, embryos were fixed 2 hr after electroporation and imaged in wholemount on a confocal microscope. The number of GFP+ cells was manually counted in the confocal z-stacks. Table 1 shows that, for a given stage, increasing the opening size of the glass capillary from 20 to 30 µm results in DNA uptake by more cells. When a single opening size was compared between stages (E7.5 versus E8.5), more cells were found to be electroporated at the latter stage. This effect may be due to a higher concentration of DNA present in the amniotic cavity at E8.5. Because the DNA solution was mixed with the green food dye, we can use the green color to assess the DNA concentration in the amniotic cavity. In the microscope, it is clear that, when compared with E8.5 embryos, the green color after DNA injection is much lighter in the cavity of E7.5 embryos. Although the same concentration of DNA solution was injected into E7.5 and E8.5 embryos, more DNA solution was in the amniotic cavity of E8.5 embryos in order to fill it completely because they are larger in the size. After withdrawing the injection needle, there is always some degree of leakage of DNA solution from the amniotic cavity, and since the puncture hole is larger in comparison to the size of the amniotic cavity in earlier embryos, it is likely that there was proportionately more leakage from E7.5 than E8.5 embryos, leading to a lower DNA concentration. The different number of transfected cells could also be due to the different diameters or induced transmembrane voltage (ITV) thresholds of cells at different stages.
A drawback of electroporation is the associated cell death. Similar to the traditional gold plated or needle-shaped electrodes, electroporation using a capillary electrode also causes the cell death. After electroporation the targeted region appeared darker in colour compared to neighbouring regions (Figure 3A and 3B), indicating that some degree of cell death must have occurred in this area. To further determine the number of dead cells caused by the electroporation procedure, embryos were stained with a fluorescent cell membrane-impermeable nuclear dye. The nuclei of dead cells were labelled with a membrane-impermeable far-red fluorescence dye. The staining confirmed that this capillary electroporation technique only results in a small number of dead cells near the electroporation site (Figure 3D and Table 1).
We noticed that although dead cells appear at the electroporation site, GFP+ cells and dead cells are also most exclusive from each other (Figure 3E and 3F). Moreover, when the caudal lateral epiblast at E8.5 was electroporated with pCAG-GFP and a glass capillary opening of 20µm, a large number of GFP+ cells was detected after 48 hr in culture (Figure 3G and 3H). Taken together, these results suggest most GFP+ cells detected 2 hr after electroporation are still viable during the further culture.
We scored the number of GFP+ cells after 24 hr ex vivo culture. Six embryos were electroporated with pCAG-GFP at E7.5, using a capillary opening of 20µm diameter. 107±31 (mean ± s.d.) GFP+ cells/embryo were detected. Since at the start of culture (2 hr), 9 cells were electroporated on average per embryo (Table 1), this suggests that electroporated cells underwent 3-4 divisions within 2 hr. The average cell doubling time from E7.5 to E8.5 embryos is around 6-7 hr in all cells apart from those in the ventral node19,20. This suggests that the electroporation procedure does not hamper normal cell growth.
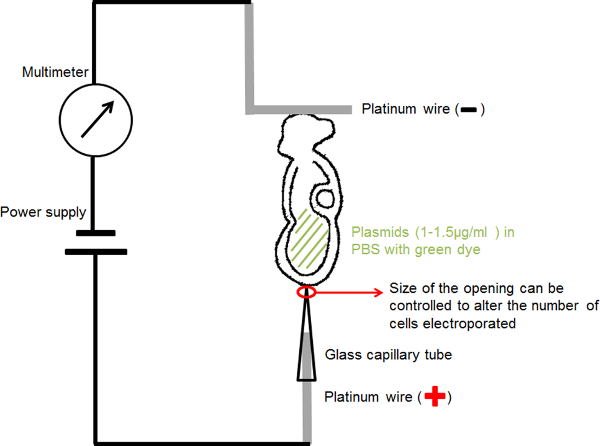
Figure 1. Circuit diagram showing the electroporation setup. The embryo containing DNA solution in its amniotic cavity was positioned between the two electrodes. Current at the chosen parameters was provided by a square wave pulse generator (power supply). A multimeter was connected in series to detect the electric current passing the embryo. Please click here to view a larger version of this figure.
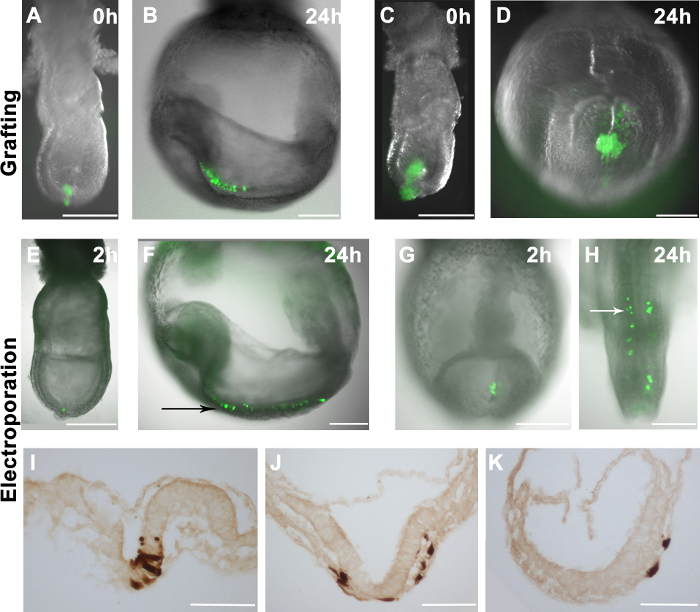
Figure 2. The distribution of grafted or electroporated cells in host embryos. (A-H) GFP fluorescence overlays (green) on brightfield images of wholemount embryos (grayscale) (A) 10-16 GFP+ EpiSCs were grafted into the distal region of a late-streak stage embryo. (C) A larger clump of GFP+ EpiSCs was grafted into the distal region of a mid-streak stage embryo. (B and D) The distribution of EpiSCs derived cells (green) in the host embryos (shown in A and C) after 24 hr in culture. (B) GFP+ cells dispersed in the host embryo, suggesting correct integration of the donor cells. (D) Grafting larger cell clumps resulted in unincorporated clump formation in the host embryo. (E-K) pCAG-Cre:GFP plasmid electroporated into specific areas of wildtype embryos. Electroporating the distal region of an early bud stage embryo (E) or the primitive streak of a 2-5 somite stage embryo (G) resulted in GFP+ cells in these regions 2 hr after the procedure. (F and H) The distribution of GFP+ cells in the host embryos after 24 hr in culture, showing that electroporated cells contribute to the neuroectoderm (black arrow) (F) and paraxial mesoderm (white arrow) (H). (I-K) DAB immunostaining for the GFP+ cells showing that the electroporated cells can give rise to the neuroectoderm (I), mesoderm (J) and endoderm (J and K) after 24 hr in culture. Scale bar (A-H) = 250 µm; scale bar (I-K) = 100 µm. Note: Figure 1A and 1B are reprinted from our previous publication4. Please click here to view a larger version of this figure.
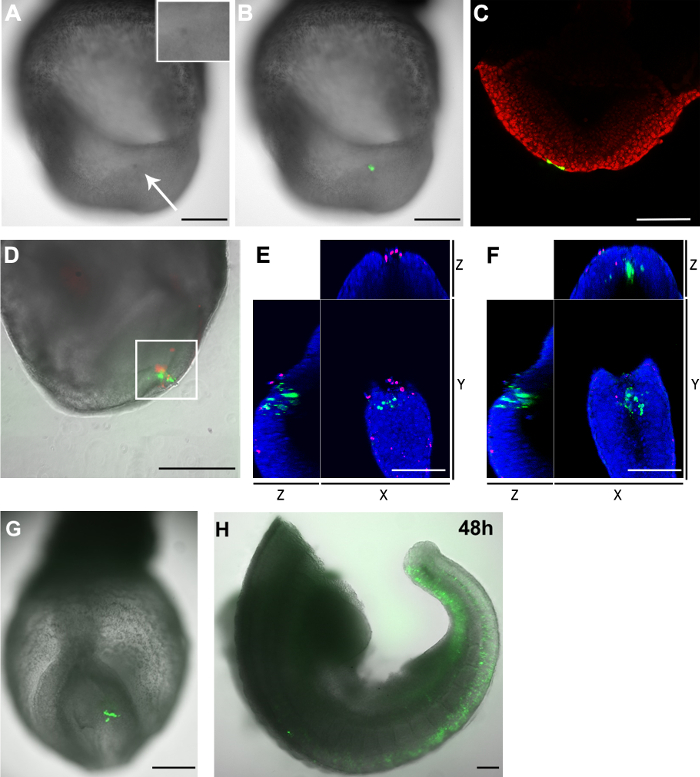
Figure 3. Distribution of GFP+ cells and dead cells in the embryos after electroporation. (A-C) pCAG-Cre:GFP plasmid electroporated into the caudal lateral epiblast cells of an E8.5 (2-5 somite stage) embryo (capillary opening size: 20 µm). (A) 2 hr after the procedure, the targeted region showed a dark color (white arrow) compared to other parts of the embryo. Inset shows an enlargement of the electroporated region. (B) Brightfield image (grayscale) overlaid with the green fluorescent channel showing the electroporated cells (green). (C) A confocal z-slice showing that two endoderm cells (green) also took up the plasmid when the caudal lateral epiblast cells were targeted. The cell nuclei are shown in red. (D-F) pCAG-Cre:GFP plasmid was electroporated in the caudal aspect of the node of an E8.5 embryo (capillary opening size: 30 µm). The embryo was cultured for 2 hr. Electroporated cells are shown in green and dead cells in red. (D) The electroporated area contains both GFP+ cells as well as dead cells. The area in the white box was further analyzed in a confocal microscope. Manual counting of the z-stack showed that there were 33 GFP+ cells and 23 dead cells in this area. Only two cells were both positive for both fluorophores. (E and F) XYZ view of a confocal z-slice from the white boxed region in D showing GFP+ cells are separate from the dead cells. The nuclei are shown in blue. (G and H) pCAG-Cre:GFP plasmid was electroporated into a few cells in the caudal lateral epiblast of an E8.5 embryo (capillary opening size: 20 µm) and imaged after two (G) and 48 (H) hours ex vivo culture Note: (H) The embryo were severed in two after culture. The head and heart regions were removed. Scale bar (A, B, D, G and H) = 250 µm; scale bar (C, E and F) = 100 µm. Please click here to view a larger version of this figure.
| Diameter of opening of the capillary tube | Embryo stage | Electroporation efficiency: no. embryos containing GFP+ cells after 2h / total no. of electroporated embryos (no. GFP+ embryos that developed normally after 24 or 48h culture) | Average number of GFP+ cells per embryo ±s.d. (n= no. of examined embryos) | No. of GFP+ endodermal cells per each embryo ±s.d. (n= no. of examined embryos) |
| 20μm | E7.5 (LS-LB) | 7 / 9 (7) | 9±3 (n=4) | 4±2 (n=4) |
| 30μm | E7.5 (LS-LB) | 13 / 15 (12) | 17±2(n=4) | 6±1 (n=4) |
| 20μm | E8.5 (2-5 somites) | 12 / 13 (10) | 21±4 (n=4) | 11±4 (n=4) |
| 30μm | E8.5 (2-5 somites) | 2 / 2 (2) | 33 and 26 (n=2) | 14 and 16 (n=2) |
Table 1. Electroporation efficiency of pCAG-Cre:GFP plasmid in mouse embryos.
Abbreviation: LS, late primitive streak stage; LB: late bud stage. Embryos are staged according to Downs and Davies12
