Efficient Nucleic Acid Extraction and 16S rRNA Gene Sequencing for Bacterial Community Characterization
Summary
We describe an efficient, robust, and cost effective method for extracting nucleic acid from swabs for characterization of bacterial communities using 16S rRNA gene amplicon sequencing. The method allows for a common processing approach for multiple sample types and accommodates a number of downstream analytic processes.
Abstract
There is a growing appreciation for the role of microbial communities as critical modulators of human health and disease. High throughput sequencing technologies have allowed for the rapid and efficient characterization of bacterial communities using 16S rRNA gene sequencing from a variety of sources. Although readily available tools for 16S rRNA sequence analysis have standardized computational workflows, sample processing for DNA extraction remains a continued source of variability across studies. Here we describe an efficient, robust, and cost effective method for extracting nucleic acid from swabs. We also delineate downstream methods for 16S rRNA gene sequencing, including generation of sequencing libraries, data quality control, and sequence analysis. The workflow can accommodate multiple samples types, including stool and swabs collected from a variety of anatomical locations and host species. Additionally, recovered DNA and RNA can be separated and used for other applications, including whole genome sequencing or RNA-seq. The method described allows for a common processing approach for multiple sample types and accommodates downstream analysis of genomic, metagenomic and transcriptional information.
Introduction
The human lower reproductive tract, gastrointestinal system, respiratory tract, and skin are colonized by complex bacterial communities that are critical for maintaining tissue homeostasis and supporting the health of the host1. For instance, certain lactobacilli create an inhospitable environment for pathogens by acidifying the vaginal vault, producing antimicrobial effectors and modulating local host immunity2-4. The growing appreciation for the bacterial microbiome's importance has also increased interest in characterizing bacterial communities in many clinical contexts. Here we describe a method to determine the composition of the bacterial microbiome from genital swabs. The protocol can be readily modified for stool samples and swabs collected from other anatomical locations and other host species.
Due to the inherent limitations in the number of samples that can be collected and stored from a given study participant, this protocol was designed to extract DNA, RNA, and potentially even protein from a single swab using an adapted phenol-chloroform based bead-beating method5,6. The combination of physical disruption of bacterial cell walls with bead-beating and chemical disruption with detergents allows rapid lysis of Gram-positive, Gram-negative, and acid-fast bacteria without additional enzymatic digestion steps. To obtain high quality RNA, it is recommended to use dry swabs that were kept at or below 4 °C immediately after collection and during transport to the laboratory (if applicable), and stored long-term at -80 °C.
To determine the bacterial microbiome within a given sample, this procedure utilizes 16S rRNA gene amplicon sequencing, which is currently the most cost-effective means to comprehensively assign bacterial taxonomy and perform relative quantification. Alternative methods include targeted qPCR7, custom microarrays8, and whole-genome sequencing9. The 16S rRNA gene contains nine hypervariable regions, and there is no consensus regarding the optimal V region to sequence for vaginal microbiome studies. Here we use the 515F/806R primer set and build on the pipeline designed by Caporaso et al.10-12. Caporaso et al.'s 515F/806R primer set enables multiplexing of hundreds of samples on a single sequencing run due to the availability of thousands of validated barcoded primers and compatibility with Illumina sequencing platforms. Unlike the Human Microbiome Project's 27F/338R primer set13, 515F/806R also effectively amplifies Bifidobacteriaceae and thus accurately captures Gardnerella vaginalis, an important member of the vaginal microbial community in some women. Alternatively, a 338F/806R primer pair has been successfully used for pyrosequencing of vaginal samples14 and a 515F/926R primer pair has recently become available for next-generation sequencing12.
Finally, this protocol provides basic instructions to perform 16S amplicon analysis using the Quantitative Insights into Microbial Ecology (QIIME) software package15. Successful implementation of the QIIME commands described here yields a table containing bacterial taxonomic abundances for each sample. Many additional quality control steps, taxonomic classification methods, and analysis steps can be incorporated into the analysis, as described in detail on the QIIME website (http://qiime.org/index.html). If the analysis will be performed on an Apple computer, the MacQIIME package16 provides easy installation of QIIME and its dependencies. Alternative software packages for 16S rRNA gene sequence analysis include Mothur17 and UPARSE18.
Protocol
The study protocol was approved by and followed the guidelines of the Biomedical Research Ethics Committee of the University of KwaZulu-Natal (Durban, South Africa) and the Massachusetts General Hospital Institutional Review Board (2012P001812/MGH; Boston, MA).
1. Extraction of Total Nucleic Acid from Cervicovaginal Swabs
Note: Perform nucleic acid extractions in sets of 16 samples or fewer. The protocol as written below assumes samples are processed in sets of 12. If performing multiple rounds of extractions, serially number the extraction batches and record each sample's extraction batch number as well as other sample information (include metadata such as the participant's ID number, age, date/time of swab collection, hormonal contraceptive type, sexually transmitted infection testing results, etc.) in Table 1.
- Preparation of reagents and fume hood
- Prepare a buffer comprised of 200 mM sodium chloride (NaCl), 200 mM Tris, and 20 mM edetic acid (EDTA) in 100 ml of nuclease-free water. Filter-sterilize the solution by passing it through a 0.22 µm filter. Chill an aliquot of 10 ml of buffer on wet ice.
- Adjust the pH of the phenol:chloroform:isoamyl alcohol (IAA) (25:24:1) to pH 7.9 by adding 65 µl of Tris alkaline buffer per 1 ml phenol, shaking the mixture for 2 min, and allowing the two phases to separate either naturally or by centrifugation at 10,000 x g for 5 min at RT.
Caution: Phenol is toxic if swallowed, if inhaled, or in contact with skin and eyes. Do not breathe fumes. Wear impervious gloves, safety glasses with side-shields, and a lab coat. - Filter-sterilize 25 ml of 20% sodium dodecyl sulfate (SDS) through a 0.22 µm filter. Make 5 ml aliquots of the sterilized SDS.
- Chill a 10 ml aliquot of isopropanol at -20 °C.
- Prepare a bead beating tube for each swab to be processed by weighing out 0.3 g of glass beads into a sterile 2 ml tube that is suitable for the bead beater.
- Obtain swabs by sampling the ectocervix with a sterile absorbent swab. Immediately after collection, place the swab into an empty and sterile cryovial, store at 4 °C for 1 to 4 hr during transport to the lab, and store for several months at -80 °C. Transfer the swabs (contained within individual tubes) to be processed to wet ice.
- Prepare the biological safety cabinet (BSC). Use a BSC with a "thimble" connected to the building exhaust to ensure proper removal of volatile chemicals.
- Remove all materials from the hood.
- Clean all surfaces of the hood with bleach, followed by a decontaminant that removes RNases, DNases, and DNA from surfaces. Clean all subsequent items brought into the hood using bleach followed by a nucleic acid decontaminant, including gloves. Use fresh RNase/DNase-free reagents, such as pipette tips, whenever possible.
- Tape a sterilized chemical biohazard bag to the rear of the hood. All dry waste containing phenol or chloroform should be placed into this bag for proper disposal.
- Place a sterile bottle into the hood to collect liquid waste containing phenol or chloroform.
- Phenol-chloroform extraction.
- In the hood, to each bead beating tube, add 500 µl of buffer (from step 1.1.1), 210 µl of 20% sodium dodecyl sulfate, and 500 µl of phenol:chloroform:IAA (25:24:1, pH 7.9).
- Transfer the swab from the transport vial into the bead beating tube using a new pair of sterile forceps. Thoroughly rub the swab head against the internal walls of the bead beating tube for at least 30 sec. Re-cap the sample when done. If performing extractions from multiple swabs, change gloves between each sample.
- Chill the sample on ice for at least 10 min. Remove the swab from the bead beating tube by holding the swab handle with sterile tweezers while pressing the swab head against the internal tube wall using a clean P200 tip. Discard the swabs in the dry chemical waste bag. Note: The "squeegee" action (pressing the swab head) will liberate liquid from the absorbent swab and increase the nucleic acid recovery.
- Place the bead beating tube into the bead beater and homogenize for 2 min at 4 °C.
- Centrifuge the bead beating tube for 3 min at 6,000 x g and 4 °C to pellet debris and separate the aqueous and phenol phases.
- Transfer the aqueous phase (~ 500 – 600 µl) to a sterile 1.5 ml tube. Add an equal volume of phenol:chloroform:IAA. Mix by inversion and brief vortexing.
- Centrifuge the tube for 5 min at 16,000 x g and 4 °C.
- Transfer the aqueous phase to a new sterile 1.5 ml tube. Be conservative and do not transfer material from the interphase layer or the underlying phenol phase. Note the volume of the transferred aqueous phase. Save the phenol phase for future protein isolation.
- Add 0.8 volume of isopropanol and 0.1 volume of 3M sodium acetate (pH 5.5). Mix thoroughly by inversion and briefly vortexing.
- Precipitate the nucleic acid by chilling the tube at -20 °C for at least 2 hr (up to O/N).
- Isopropanol precipitation and ethanol wash
- Centrifuge the tube for 30 min at approximately 16,000 x g and 4 °C. Carefully use a pipette to remove the supernatant, leaving the pellet intact.
- Add 500 µl of 100% ethanol. Dislodge the pellet with gentle vortexing or pipetting without touching the pellet. Centrifuge for 5 min at 16,000 x g and 4 °C.
- Carefully discard the ethanol supernatant. Use a P10 pipet to remove as much ethanol as possible without disturbing the pellet.
- Air dry the pellet at RT for 15 min.
- Resuspend the pellet in 20 µl of ultra-pure 0.1x Tris-EDTA buffer. Allow the sample to chill on ice for 10 min and pipette repeatedly to ensure full resuspension. If the pellet does not dissolve, transfer the tube to a 40 °C heat block for up to 10 min to aid dissolution.
- Measure the nucleic acid concentration using a spectrophotometer19.
- If desired, separate DNA from RNA using a column clean-up kit, following the manufacturer's protocol20.
- Store the nucleic acid at -80 °C or continue.
2. PCR Amplification of the 16S rRNA Gene V4 Hypervariable Region
Note: Perform the PCR amplification in sets of 12 samples or fewer to minimize the risk of contamination and human error. If performing multiple rounds of amplification, serially number the amplification batches and record each sample's amplification batch number in Table 1.
- Preparation of the reagents and PCR hood
- Add the PCR amplification set information to Table 1, which will serve as the basis of the mapping file at the sequence analysis stage.
- Remove all materials from a PCR hood and clean the internal surfaces thoroughly with bleach followed by a decontaminant that removes RNases, DNases, and DNA. Be sure to decontaminate every reagent and piece of equipment (e.g., pipettes) before placing them in the hood. Wear fresh gloves cleaned with a nucleic acid decontaminant prior to working in the hood.
- If necessary, dilute the nucleic acid template to 50 – 100 ng/µl using DNA-free and nuclease-free water.
- Thaw aliquots of the 5x high-fidelity (HF) buffer, dNTPs, and primers in the clean PCR hood. Gently vortex and centrifuge all solutions after thawing. To minimize freeze-thaw cycles and the risk of stock contamination, prepare aliquots of the 5x HF buffer, dNTPs, and primers.
- Place a clean benchtop cooler rack for microcentrifuge tubes and a PCR plate cooler into the hood.
- For PCR reaction, prepare the master mix by combining 15.5 µl of ultra-pure water, 5 µl of 5x HF buffer, 0.5 µl of dNTPs, 0.5 µl of 515F forward primer, 0.75 µl of 3% DMSO, and 0.25 µl of Polymerase for each reaction. Assemble all reaction components in the cooler and add the polymerase last. Mix thoroughly by pipetting. Add two extra samples to the reaction count when preparing the master mix, to account for pipetting error.
- PCR reaction setup:
Note: Perform amplifications in triplicate, meaning each sample is amplified in three separate 25 µl reactions. Run a no-template water control with each primer pair. Work quickly but carefully, avoiding introduction of any contamination.- Label an 8-well strip with individual caps and place into a PCR cooler.
- Pipette 90 µl of master mix into the first well.
- Add 2 µl of the reverse primer (Supplemental File 1). Be sure to carefully note the reverse primer barcode used with each sample in Table 1.
- Mix well and transfer 23 µl of master mix to the fourth well (the no-template control).
- Add 2 µl of water to the fourth well.
- Add 6 µl of the appropriate sample to the first well. Mix well and transfer 25 µl to the second well. Change tips and transfer another 25 µl from the first well to the third well. Firmly cap every well, making sure not to touch the inside of the wells or cap in the process.
- Repeat for each sample.
- Perform PCR amplification
- Transfer the strip tubes to a thermocycler and run the following program: 30 sec at 98 °C, followed by 30 cycles of 10 sec at 98 °C, 30 sec at 57 °C, and 12 sec at 72 °C, followed by a 10 min hold at 72 °C and final hold at 4 °C.
- Perform the following steps on a clean lab bench. Quickly spin the tubes to collect liquid from the walls. Combine triplicate PCR reactions from each sample, with a total volume of 75 µl, into a sterile labeled tube. Also transfer 25 µl of each no-template control into a separate sterile tube. Do not combine amplicons from different samples yet.
- Validation of successful PCR amplification of samples by gel electrophoresis.
- Prepare a 1.5% agarose gel (1.5 g agarose powder in 100 ml of 1x TAE buffer) with enough wells to hold each amplicon, water control, and ladder21.
- While the gel hardens (about 30 min), prepare the sample for electrophoresis: Add 1 µl of 6x loading dye to a new, labeled tube. To that tube, add 5 µl of the amplicon and mix by pipetting.
- When the gel has set, remove the combs, place the gel in the electrophoresis tank, and fill the tank with 1x TAE buffer.
- To the first well, add 5 µl of DNA ladder.
- Load 5 µl of the sample amplicon to another well. Load 5 µl of the no-template amplicon to a separate well. Continue as needed for each sample.
- When all samples have been loaded, slide the tank lid in place and turn on the power source to 120 V. Allow the gel to run for 30 – 60 min.
- View the gel under UV light.
- Verify successful amplification of each sample by noting a single strong band around 380 bp. If there is a double band, re-amplify the sample with a different reverse barcode (Step 2.3). If there is no band at all, re-amplify the sample using either the same reverse barcode or a new reverse barcode (Step 2.3). If re-amplification is unsuccessful, PCR inhibitors may be present in the sample, in which case, perform a column-based DNA cleanup to remove PCR inhibitors.
Note: Successful amplification may not be possible if the bacterial DNA concentration in the original sample is insufficient (<5 ng/µl). - Verify the lack of reagent contamination by noting the absence of a band in the no-template control.
- Verify successful amplification of each sample by noting a single strong band around 380 bp. If there is a double band, re-amplify the sample with a different reverse barcode (Step 2.3). If there is no band at all, re-amplify the sample using either the same reverse barcode or a new reverse barcode (Step 2.3). If re-amplification is unsuccessful, PCR inhibitors may be present in the sample, in which case, perform a column-based DNA cleanup to remove PCR inhibitors.
- Store the remaining 70 µl of amplicon at -20 °C. Discard the remaining 20 µl of the no-template control, assuming it did not yield a band.
3. Library Pooling and High-Throughput Sequencing
- Create the amplicon pool by combining an equal volume (2 – 5 µl) of each amplicon into a single sterile tube. If the band from a sample looked particularly weak, add twice the volume relative to the rest of the samples.
- Remove the PCR primers from the amplicon pool using a PCR Clean-up kit, following the manufacturer's instructions22. Perform the clean-up with multiple columns if the amplicon pool volume is over 100 µl. Note: Each column has a 100 µl capacity.
- Store the library at -20 °C or proceed to the next step.
- If applicable, combine the primer-free amplicon pools to create the final library. Determine the DNA concentration of the library using a spectrophotometer or a fluorometric system23. A 260/280 ratio between 1.8 – 2.0 is indicative of pure DNA.
- Dilute the library to 20 nM. Confirm the quality of the library by visualizing a single band around 400 bp using an electrophoresis instrument. Confirm the concentration of the library using a fluorometric system23.
- Perform a final 1:10 dilution in water to dilute the library to 2 nM. Then, store the library at 20 °C indefinitely.
- Send an aliquot of the final library with the three required sequencing primers (Read 1, Read 2, and Index; see Tables of Materials/Equipment) to be sequenced on an Illumina sequencer. If fewer than 300 samples have been multiplexed for sequencing, use a single-end 300 bp run and with a 12 bp index read on a MiSeq, with a final library concentration of 5 pM and a 10% denatured PhiX spike-in. See the supplemental materials of Caporaso et al. ISME J, 201210 for detailed sequencing instructions.
4. Sequence Analysis
Note: Outlined here is a basic pipeline for sequence analysis using the QIIME 1.8.0 software package. For simplicity, the provided commands assume that the mapping file is called mapping.txt, the 12 bp index read file is called index.fastq, and the 300 bp sequencing read file is called sequences.fastq. Install QIIME or MacQIIME16 and familiarize yourself with the basics of UNIX to execute these commands. Read the complete guide to QIIME at:
- Complete the mapping file for the experiment (Table 1). Include as much metadata as possible. Note which samples have been extracted or amplified in the same batch, to determine whether there are batch effects.
- Save the mapping file as a text file, e.g., mapping.txt. Validate the formatting of the mapping file by executing the following command:validate_mapping_file.py -m mapping.txt -o mapping_output
Note: This command uses the built-in "validate_mapping_file.py" QIIME script that makes a new folder, called "mapping_output", containing an .html file indicating the mapping file errors, if any. - Check the quality of the sequencing reads using a high-throughput sequence data quality checking program, such as FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Figure 5 demonstrates the per base sequence quality that can be expected from a successful run.
Note: The sequencer assigns each nucleotide base a Phred quality score, which corresponds to the probability that the base has been erroneously called. A Phred quality score of 10 indicates that there is a 10% chance that the nucleotide has been incorrectly assigned, 20 indicates a 1% chance, 30 indicates a 0.1% chance, and 40 (the highest possible score) indicates a 0.01% chance24. - Using the mapping file as a key, demultiplex, quality filter the sequencing data, and save the results to a folder (in this case, called "sl_out") by executing this command25: split_libraries_fastq.py –rev_comp_mapping_barcodes -i sequences.fastq -o sl_out/ -b index.fastq -m mapping.txt -q 29
Note: The q flag denotes the maximum unacceptable Phred quality score, e.g., "-q 29" filters out any sequences with Phred scores below 30, ensuring 99.9% accuracy of the base calls. - Using the Greengenes 16S operational taxonomic unit (OTU) reference database26 (http://qiime.org/home_static/dataFiles.html), perform open-reference OTU picking by executing this command27: pick_open_reference_otus.py -i sl_out/seqs.fna -r 97_otus.fasta -o ucrss/ -s 0.1
Note: The -s flag indicates the fraction of sequences that failed to align to the reference database that will be included in the de novo clustering. "-s 0.1" includes 10% of the failed sequences in the de novo clustering. Use the -a flag to parallelize the OTU picking process and reduce the processing time from days to hours if multiple cores are available. - Create a user-friendly taxonomic abundance table by merging OTUs at the species level by executing this command28: summarize_taxa.py -i ucress/otu_table_mc2.biom -o summarized_otuSpecies/ -L 7
Note: The resulting table can be easily viewed in any spreadsheet software. Note that 16S rRNA sequencing does not reliably provide species level resolution. - Determine the ecological diversity within each sample by computing several alpha diversity metrics with the QIIME script alpha_diversity.py. Then, determine the diversity between pairs of samples using the QIIME script beta_diversity.py.
- Visualize the data, e.g., by using an EMPeror29 principal coordinates plot or heatmap.
- Perform formal statistical comparisons of mapping file categories, e.g., with QIIME's compare_catagories.py script30.
Representative Results
The general overview of the protocol, which enables the determination of relative bacterial abundances from a swab using 16S rRNA gene sequencing, is shown in Figure 1.The protocol has been optimized for human vaginal swabs, but can be easily adapted for most mucosal sampling sites and other hosts. Figure 2 demonstrates the high-quality DNA and RNA that can be isolated using the bead-beating protocol. Figure 3 illustrates a successful PCR amplification of 12 samples, where each amplification with a sample yielded a single strong band of the correct size and each water control did not yield a band. Figure 4 illustrates the quantification of the final library pool prior to sequencing. Figure 5 shows a typical sequence quality profile after a single-end 300 bp MiSeq run.

Figure 1. Schematic Overview of the Protocol. First, nucleic acid is extracted from a swab by bead-beating in a buffered solution containing phenol, chloroform, and isoamyl alcohol. Variable region 4 of the 16S rRNA gene is then amplified from the resulting nucleic acid using PCR. PCR amplicons from up to hundreds of samples are then combined and sequenced on a single run. The resulting sequences are matched to a reference database to determine relative bacterial abundances. The entire protocol can be performed in approximately three days. Please click here to view a larger version of this figure.
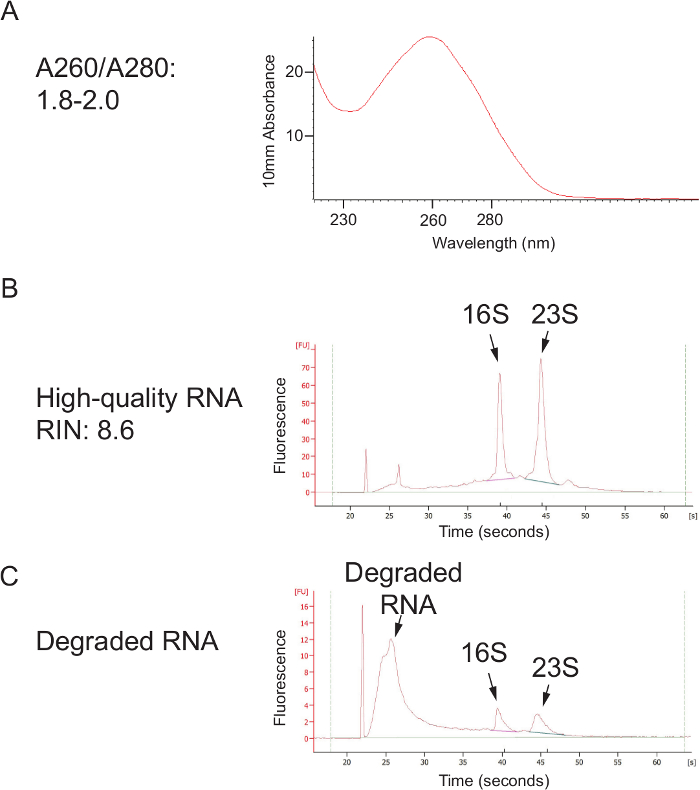
Figure 2. High-quality Nucleic Acid Extracted Using the Phenol:Chloroform Bead Beating Method. (A) DNA quality, as assessed using a spectrophotometer. An A260/A280 ratio between 1.8 and 2.0 indicates pure nucleic acid that is not contaminated with phenol or protein. (B) After a column clean-up, this protocol can yield high-quality RNA, indicated by strong 16S and 23S rRNA peaks. (C) RNA degradation can occur if the sample is not kept cold after collection (during transport and storage) or if RNases are present during processing. Please click here to view a larger version of this figure.
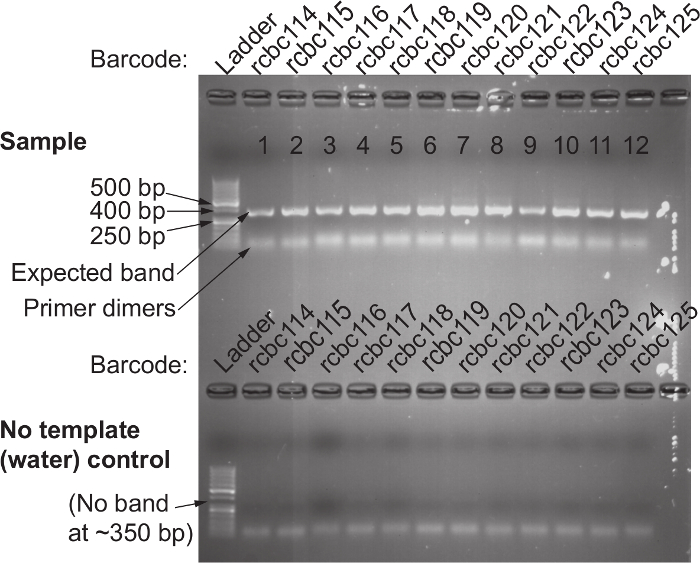
Figure 3. Confirmation of Successful 16S rRNA Gene Amplification Using the 515F and Barcoded 806R Primer Set. Top) Gel electrophoresis is used to confirm the presence of a single band around 380 base pairs in every sample that was amplified with template. The absence of a band indicates unsuccessful amplification; this is usually due to human error and the PCR reaction from that sample should be repeated. Bottom) No template (water) controls run in parallel with the same primer pair should not have a band present. The presence of a band in the water control indicates contaminated reagents; discard the reagents that may be contaminated and re-do the PCR amplifications of both the template and water control for that primer pair. Please click here to view a larger version of this figure.
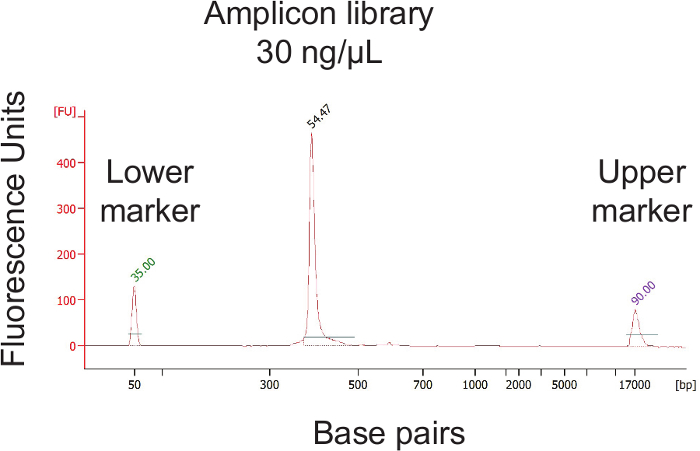
Figure 4. Quantification of the Final Library Pool Concentration and Validation of the Library Size. After pooling the individual sample amplicons, the concentration of the final library pool must be determined. The library pool must then be further diluted to achieve a 2 nM concentration. Please click here to view a larger version of this figure.
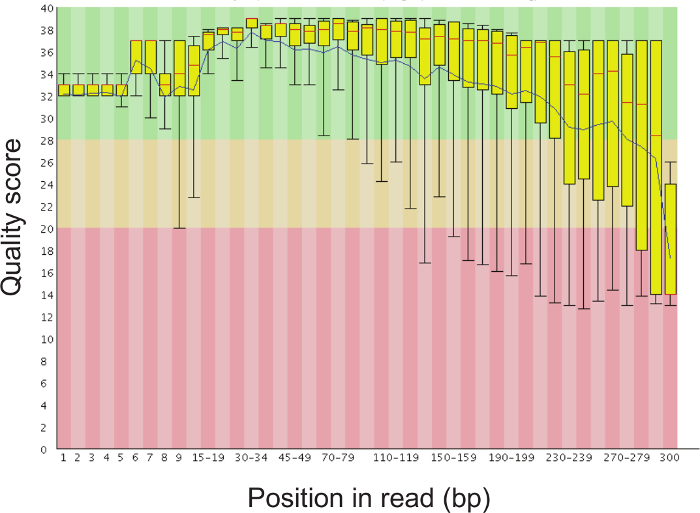
Figure 5. Representative Bar Plot of the Sequence Quality Scores at Each Position of the Read. It is normal for the sequence quality to drop after 200 base pairs, but the average quality score should remain above 30. Please click here to view a larger version of this figure.
| #SampleID | Barcode Sequence |
LinkerPrimer Sequence |
rcbcPrimer | SampleType | Extraction Batch |
Amplification Plate |
Description | |||
| #An example mapping file can be found at: http://qiime.org/_static/Examples/File_ Formats/ Example_ Mapping_ File.txt |
||||||||||
| AG2350 | TCCCTTGTCTCC | CCGGACTACHVGGGTWTCTAAT | rcbc000 | Cervical Swab |
1 | A | ||||
Table 1. Mapping File Template. Creating an accurate and thorough mapping file is critical for successfully executing the protocol. The mapping file is not only required for executing QIIME, but it also enables the researcher to maintain the link between the sample barcode and metadata, to analyze the data for any systematic biases (e.g., batch-to-batch variation), and to determine interesting correlations between the metadata and bacterial populations. A bare-bones mapping file is provided, but users are encouraged to add as many columns containing metadata as possible. Examples of additional metadata for a vaginal swab includes the participant's age, date/time of swab collection, hormonal contraceptive type (if applicable), sexually transmitted infection testing results, etc.
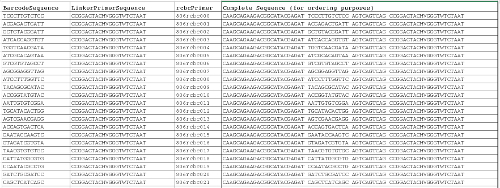
Supplemental File 1. List of Barcoded Reverse Primer Sequences10. The first three columns can be used to complete the mapping file, and the last column provides the entire primer sequence for ordering purposes. Please click here to download this file.
Discussion
Here we describe a protocol for the identification and characterization of relative bacterial abundances within a human vaginal swab. This protocol can easily be adapted for other sample types, such as stool and swabs of other body sites, and for samples collected from a wide variety of sources. The extraction of nucleic acid by bead-beating in a buffered solution of phenol and chloroform allows for isolation of both DNA and RNA, which is particularly important when working with precious samples collected through clinical studies. The isolated bacterial DNA is excellent for bacterial taxonomic identification and genomic assembly, while the simultaneous collection of RNA provides the opportunity to determine functional bacterial, host, and viral contributions through RNA-seq. The described protocol uses a validated one-step primer set that has been successfully deployed on a wide range of sample types, including human, canine, and environmental samples10. The availability of thousands of barcoded primers enables multiplexing of samples and tremendous savings on sequencing costs. The complete cost (including all reagents, a single sequencing run, and primers but not equipment) is about $20 per sample when 200 samples are multiplexed. Additionally, there is very high reproducibility when multiple swabs from the same sample site are processed independently through the entire pipeline. Overall, the protocol is cost efficient, flexible, reliable, and repeatable.
The nucleic acid extraction portion of this protocol is limited by the safety precautions required when working with phenol and chloroform, and the challenges of automating the pipeline to a high-throughput, 96-well plate format. Additionally, the vigorous bead beating used for mechanical lysis shears the bacterial DNA to approximately 6 kilobase fragments; if longer DNA fragments are required for downstream applications, the duration of bead beating should be shortened. The limitations of the bacterial identification portion of this protocol are inherent to any method that relies on 16S rRNA gene sequencing. 16S rRNA sequencing is ideal for bacterial identification to the genus and even species level, but rarely provides strain level identification. While the V4 variable region of the 16S rRNA gene provides robust discrimination amongst most bacterial species11, additional computational methods such as Oligotyping31 may need to be used to precisely identify certain species, such as Lactobacillus crispatus. Finally, information about the precise bacterial functional capabilities within a particular sample cannot be determined by 16S rRNA gene sequencing alone, though this protocol enables extraction of whole genome DNA and RNA that can be used towards this purpose.
The most critical step to ensuring success with this protocol is taking great care to prevent contamination during sample collection, nucleic acid extraction, and PCR amplification. Ensure sterility at the time of sample collection by wearing clean gloves and using sterile swabs, tubes, and scissors. To assess for contamination of the collection materials, collect negative control swabs by placing additional unused swabs directly into transport tubes at the time of sampling. In the lab, perform all pre-amplification steps in a sterilized hood containing only decontaminated supplies and using only molecular grade, DNA-free reagents. During nucleic acid extraction, prevent cross-contamination by using new sterile forceps and fresh gloves with each sample, and keeping all tubes closed unless in use. Processing unused swabs in parallel ensures sterility of both the sample collection and nucleic acid extraction; the unused swabs should not yield a pellet after isopropanol precipitation and ethanol washing. If a pellet does appear, perform 16S rRNA gene amplification to determine a possible source of the contamination (e.g., the presence of Streptococcus or Staphylococcus would indicate skin contamination). Additionally, perform PCR amplifications with no template control reactions in parallel to ensure that the PCR reagents and reactions have not been contaminated. If a band appears in a no template control, discard the reagents and repeat the amplification with fresh reagents. Taking these precautions will ensure successful sequencing of the bacteria of interest.
The PCR amplification step tends to require the most troubleshooting. Amplifying in sets of twelve samples provides a balance between efficiency and consistency. The complete absence of bands across all samples in a given amplification set indicates a systematic failure, e.g., forgetting to add a reagent or incorrectly programming the thermocycler. The absence of a band from a few samples is usually due to human error, and the amplifications should be re-run with the same pairing of sample and reverse primer. In the case of continued absence of a band, the sample can be re-amplified using a reverse primer with a different barcode. Repeated amplification failures with multiple reverse primers may indicate an inhibitor present in the sample. In that case, cleaning the DNA with a column will often remove inhibitors without significantly altering relative bacterial abundances. If multiple bands result after amplification, re-amplify the sample with a different reverse primer barcode.
In addition to preventing environmental contamination and ensuring amplification of a single specific product, successful sequencing relies on care when preparing the library pool. The goal is to combine equimolar amounts of each sample's amplicons to ensure approximately the same number of sequencing reads per sample. If the nucleic acid concentrations prior to amplification are comparable, simply adding equal volumes of each sample's amplicons is sufficient when creating the library pool. However, if the nucleic acid concentrations are vastly different and added in equal volume, the sample with the low nucleic acid concentration will be poorly represented with a low number of reads. In this case, it is possible to add a higher volume of the amplicons from the low concentration sample based upon the relative intensity of the gel band. Alternatively, it is possible to more rigorously remove primers from the individual amplicons, quantify individual sample's amplicon concentration using a fluorometric dsDNA quantification kit, and precisely combine equimolar amounts of each sample.
Once a well-balanced amplicon pool is generated, it becomes critical to carefully measure the pool's concentration. Subsequent careful dilution and spike-in with PhiX to increase the read complexity is critical for achieving optimal sequencing results. High-throughput sequencers that use sequencing by synthesis are very sensitive to the cluster density on the flow cell. Loading a library pool that is too concentrated will result in overclustering, with lower quality scores, lower data output, and inaccurate demultiplexing32. Loading a library pool that is too dilute will also result in low data output. Carefully quantifying the library pool prior to sequencing will ensure optimal results.
16S rRNA gene sequencing provides a comprehensive assessment of the bacteria present within a given sample and is an absolutely critical first step in hypothesis generation. The presence of a rich set of metadata further enables the researcher to test associations between particular bacterial species and important biological factors. Furthermore, the same 16S information can be used to infer the bacterial functions using with tools such as PICRUSt33. The ultimate goal is to use 16S characterization to identify novel associations that can be further tested and validated in model systems, adding to our growing understanding of the impact of the bacterial microbiome on human health and disease.
Declarações
The authors have nothing to disclose.
Acknowledgements
We would like to thank Elizabeth Byrne, David Gootenberg, and Christina Gosmann for critical feedback on the protocol; Megan Baldridge, Scott Handley, Cindy Monaco, and Jason Norman for sample preparation guidance and demonstrations; Wendy Garrett, Curtis Huttenhower, Skip Virgin, and Bruce Walker for protocol advice and fruitful discussions; and Jessica Hoisington-Lopez for sequencing support. This work was supported by the Bill and Melinda Gates Foundation and the NIAID (1R01AI111918). D.S.K. received additional support from the Burroughs Wellcome Fund. M.N.A. was supported by award number T32GM007753 from the NIGMS, and the Paul and Daisy Soros Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the NIH.
Materials
| Equipment: | |||
| Mini-Beadbeater-16 | BioSpec | 607 | |
| PCR workstation | Any PCR hood can be used, e.g., the AirClean 600. | ||
| Thermocycler | Any thermal cycler with a heated lid can be used, e.g., MJ Research PTC-200. | ||
| Electrophoresis system | Any electrophoresis system can be used, e.g. the Thermo Scientific Owl EasyCast B1 Mini Gel Electrophoresis system. | ||
| Nanodrop | Thermo Scientific | 2000C | Any other DNA quantification method will be sufficient |
| Bioanalyzer | Agilent | 2100 | An alternative is the Agilent 2200 TapeStation Instrument. Not absolutely necessary but very helpful. |
| MiSeq or HiSeq | Illumina | ||
| Name | Company | Catalog Number | Comments |
| Materials: | |||
| Catch-All Sample Collection swab | Epibio | QEC89100 | Other swabs can be used but the Catch-All swab is recommended by the Human Microbiome Project. |
| ELIMINase | Fisher | 04-355-31 | |
| SteriFlip 50 mL filtration device (0.22 µm) | EMD Millipore | SCGP00525 | |
| 0.1 mm glass beads | BioSpec | 11079101 | |
| 2 mL screw-cap tubes | Sarstedt | 72.694.006 | For bead beating |
| UltraPure 5M NaCl | Life Technologies | 24740-011 | Molecular Biology Grade |
| 1 M Tris-HCl | Ambion (Invitrogen) | AM9856 | Molecular Biology Grade |
| 0.5 M EDTA | Ambion (Invitrogen) | AM9260G | Molecular Biology Grade |
| Sodium Dodecyl Sulfate, 20% Solution | Fisher | BP1311-200 | Molecular Biology Grade |
| UltraPure DNase/RNase-free distilled water | Ambion | 10977-015 | Molecular Biology Grade, for buffer preparation |
| 2-Propanol BioReagent, for molecular biology, ≥99.5% | Sigma | I9516-500ML | Molecular Biology Grade |
| Phenol:Chloroform:IAA, 25:24:1 | Invitrogen | AM9730 | Warning: Toxic |
| 3 M Sodium Acetate, pH 5.5 | Life Technologies | AM9740 | Molecular Biology Grade |
| Disposable sterile polystyrene forceps, PS | Cole Parmer | EW-06443-20 | |
| 1.5 mL, clear, PCR clean tubes | Eppendorf | 22364120 | |
| PCR grade water | MoBio | 17000-11 | For PCR |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | M0530S | |
| dNTP mix | Sigma | D7295-0.5mL | |
| 0.2 ml PCR 8-tube with attached clear flat caps, natural | USA Scientific | 1492-3900 | Any 8-tube strips that are DNase, RNase, DNA, and PCR inhibitor free will work |
| Agarose | BioExpress | E-3121-25 | |
| 50X TAE buffer | Lonza | 51216 | |
| DNA gel stain | Invitrogen | S33102 | |
| 6X DNA Loading Dye | Thermo (Fisher) | R0611 | |
| 50bp GeneRuler Ladder | Thermo (Fisher) | SM0373 | |
| AllPrep DNA/RNA kit | Qiagen | 80284 | |
| UltraClean PCR Clean-up Kit | MoBio | 12500-100 | |
| Quant-iT PicoGreen dsDNA Assay Kit | Thermo Fisher Scientific | P11496 | An alternative is Qubit Fluorometric Quantification (Life Technologies) |
| Name | Company | Catalog Number | Comments |
| Primers: | |||
| 515F (forward primer) 5'-AATGATACGGCGACCACCGAG ATCTACACTATGGTAATTGT GTGCCAGCMGCCGCGGTAA-3' |
Order at 100 nmole; Purification: Standard Desalting. Resuspend at 100 µM. **Critical: primers must be resuspended with MoBio PCR Grade Water (see above) in a hood to avoid contamination.** | ||
| Reverse primers, see the Supplemental Code File and: ftp://ftp.metagenomics.anl.gov/data/misc/EMP/SupplementaryFile1_barcoded _primers_515F_806R.txt |
IDT is recommended | If ordering large sets of primers, order as a 96-well plate at the 100 nmole scale. Resuspend at 100 μM. Full directions for primer ordering and resuspension at http://www.earthmicrobiome.org/files/2013/04/EMP_primer_ordering_and _resuspension.doc. **Critical: primers must be resuspended with MoBio PCR Grade Water (see above) in a hood to avoid contamination.** |
|
| Read 1 Sequencing Primer 5'-TAT GGT AAT TGT GTG CCA GCM GCC GCG GTA A-3' | 25 nmole; Purification: Standard Desalting. Resuspend at 100 µM. | ||
| Read 2 Sequencing Primer 5'-AGT CAG TCA GCC GGA CTA CHV GGG TWT CTA AT-3' | 26 nmole; Purification: Standard Desalting. Resuspend at 100 µM. | ||
| Index Sequencing Primer 5'-ATT AGA WAC CCB DGT AGT CCG GCT GAC TGA CT-3' | 27 nmole; Purification: Standard Desalting. Resuspend at 100 µM. | ||
| PhiX Control v3 | Illumina | FC-110-3001 | Required if performing the sequencing in-house. If the sequencing will be performed by a third-party sequencing center, they will already have PhiX. |
Referências
- Huttenhower, C. Structure, function and diversity of the healthy human microbiome. Nature. 486, 207-214 (2012).
- O’Hanlon, D. E., Moench, T. R., Cone, R. A. Vaginal pH and microbicidal lactic acid when lactobacilli dominate the microbiota. PloS one. 8, e80074 (2013).
- Aldunate, M. Vaginal concentrations of lactic acid potently inactivate HIV. The Journal of antimicrobial chemotherapy. 68, 2015-2025 (2013).
- Anahtar, M. N., et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity. 42, 965-976 (2015).
- Reyes, A., Wu, M., McNulty, N. P., Rohwer, F. L., Gordon, J. I. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proceedings of the National Academy of Sciences of the United States of America. 110, 20236-20241 (2013).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical biochemistry. 162, 156-159 (1987).
- Srinivasan, S., et al. Temporal variability of human vaginal bacteria and relationship with bacterial vaginosis. PloS one. 5, e10197 (2010).
- Dols, J. A., et al. Microarray-based identification of clinically relevant vaginal bacteria in relation to bacterial vaginosis. American journal of obstetrics and gynecology. 204, 301-307 (2011).
- Segata, N., et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature methods. 9, 811-814 (2012).
- Caporaso, J. G., et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal. 6, 1621-1624 (2012).
- Caporaso, J. G., et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America. 108, 4516-4522 (2011).
- Ravel, J., et al. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America. 108, 4680-4687 (2011).
- Srinivasan, S., et al. Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PloS one. 7, e37818 (2012).
- Caporaso, J. G., et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 7, 335-336 (2010).
- Schloss, P. D., et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 75, 7537-7541 (2009).
- Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature. 10, 996-998 (2013).
- AllPrep DNA/RNA Mini Kit. Qiagen Available from: https://http://www.qiagen.com/us/shop/sample-technologies/rna-sample-technologies/dna-rna-protein/allprep-dnarna-mini-kit/ (2015)
- Lee, P. Y., Costumbrado, J., Hsu, C. Y., Kim, Y. H. Agarose gel electrophoresis for the separation of DNA fragments. Journal of visualized experiments. , (2012).
- Quant-iT PicoGreen dsDNA Reagent. Invitrogen Available from: https://tools.thermofisher.com/content/sfs/manuals/mp07581.pdf (2008)
- DeSantis, T. Z., et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 72, 5069-5072 (2006).
- Vazquez-Baeza, Y., Pirrung, M., Gonzalez, A., Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. GigaScience. 2, 16 (2013).
- Eren, A. M., et al. Exploring the diversity of Gardnerella vaginalis in the genitourinary tract microbiota of monogamous couples through subtle nucleotide variation. PloS one. 6, e26732 (2011).
- Langille, M. G., et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 31, 814-821 (2013).
