التسلسل والدراسات التنميط الجيني أساس، بما في ذلك دراسات الجينوم على نطاق الرابطة (GWAS)، دراسات المرشح مكان، وأعماق التسلسل الدراسات، وقد حددت العديد من المتغيرات الجينية التي ترتبط إحصائيا مع المرض، وسمة، أو النمط الظاهري. وخلافا للتوقعات في وقت مبكر، وتقع معظم هذه المتغيرات (85-93٪) في مناطق غير الترميز ولا تغيير تسلسل الأحماض الأمينية للبروتينات 1،2. تفسير وظيفة من هذه المتغيرات غير الترميز وتحديد الآليات البيولوجية وربطها لمرض المرتبطة بها، وقد ثبت سمة، أو النمط الظاهري تحديا 3-6. لقد قمنا بتطوير استراتيجية عامة لتحديد الآليات الجزيئية التي تصل المتغيرات إلى النمط الظاهري وسيط مهم – التعبير الجيني. تم تصميم هذا الخط على وجه التحديد لتحديد التشكيل من فريق العمل ملزم المتغيرات الجينية. هذه الاستراتيجية يجمع بين النهج الحسابية وتقنيات البيولوجيا الجزيئية التي تهدف إلى التنبؤالآثار البيولوجية من المتغيرات مرشح في سيليكون، وتحقق هذه التنبؤات تجريبيا (الشكل 1).
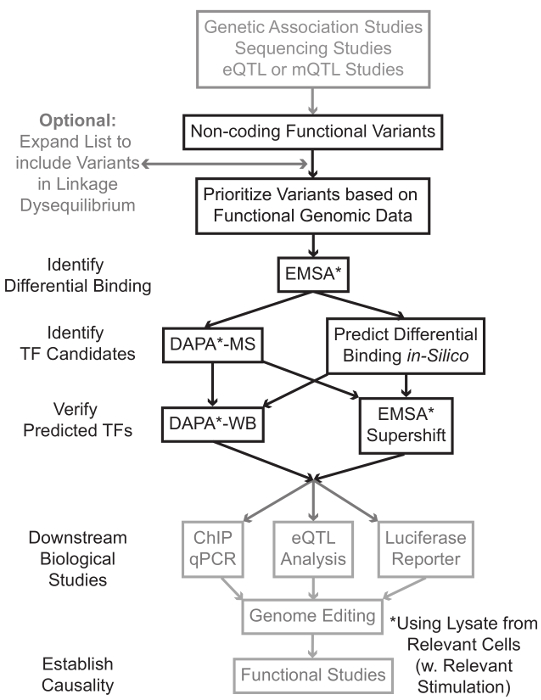
الشكل 1: النهج الاستراتيجي لتحليل خطوات غير ترميز المتغيرات الجينية التي لم يتم تضمينها في بروتوكول مفصلة المرتبطة مظللة باللون الرمادي هذا المخطوط الرجاء انقر هنا لعرض نسخة أكبر من هذا الرقم.
في كثير من الحالات، من المهم أن تبدأ من خلال توسيع قائمة المتغيرات بحيث يشمل جميع من في ارتفاع الربط-اختلال التوازن (LD) مع كل متغير يرتبط إحصائيا. LD هو مقياس لجمعية غير عشوائية من أليل على موقعين الكروموسومات المختلفة، والتي يمكن أن تقاس ص 2 الإحصائية 7. ص 2 هو مقياس للينكاجي اختلال التوازن بين الخيارين، مع ص 2 = 1 تدل على الربط المثالي بين الخيارين. تم العثور على الأليلات في LD عالية للمشاركة في فصل على كروموسوم عبر السكان الأسلاف. لا تشمل صفائف التنميط الجيني الحالية كافة المتغيرات المعروفة في الجينوم البشري. بدلا من ذلك، فإنها تستغل LD داخل الجينوم البشري وتشمل مجموعة فرعية من المتغيرات المعروفة التي تعمل كوكلاء للالمتغيرات الأخرى داخل منطقة معينة من LD 8. وهكذا، وهو البديل من دون أي نتيجة البيولوجية قد تترافق مع مرض معين لأنه في LD مع السببية البديل، البديل مع التأثير البيولوجي ذات مغزى. من الناحية الإجرائية، فمن المستحسن لتحويل الإصدار الأخير من 1000 الجينوم المشروع 9 ملفات دعوة البديل (VCF) في الملفات الثنائية متوافقة مع طقطقة 10،11، أداة مفتوحة المصدر للتحليل جمعية الجينوم بأكمله. وفي وقت لاحق، كل المتغيرات الجينية الأخرى مع LD ص 2> 0.8 مع كل فا الجيني المدخلاتويمكن تحديد RIANT كمرشحين. ومن المهم استخدام السكان إشارة المناسب لهذا خطوة- على سبيل المثال، إذا تم تحديد البديل في المواد من أصول أوروبية، وينبغي استخدام البيانات من موضوعات من أصل مماثل للتوسع دينار.
التوسع LD غالبا ما يؤدي إلى العشرات من المتغيرات مرشح، وأنه من المرجح أن جزءا صغيرا فقط من هذه تسهم في آلية المرض. في كثير من الأحيان، أصبح في حكم المستحيل لدراسة التجربة كل من هذه المتغيرات بشكل فردي. ولذا فمن المفيد الاستفادة من آلاف من مجموعات البيانات الجينومية الوظيفية المتاحة علنا كمرشح لتحديد أولويات المتغيرات. على سبيل المثال، اتحاد ترميز 12 قد أنجز الآلاف من تجارب رقاقة وما يليها واصفا ربط TFS والعوامل المشتركة، وعلامات هيستون في مجموعة واسعة من السياقات، جنبا إلى جنب مع البيانات لونين الوصول من التقنيات مثل الدناز تسلسل 13، أي تي أي سي -seq 14، وFAIRE وما يليها 15. DatabASES وخوادم الويب مثل متصفح UCSC الجينوم 16، خارطة الطريق Epigenomics 17، مخطط Epigenome 18، Cistrome 19، وإعادة رسم خريطة 20 توفر حرية الوصول إلى البيانات التي تنتجها هذه وتقنيات تجريبية أخرى عبر مجموعة واسعة من أنواع الخلايا والشروط. عندما يكون هناك الكثير من المتغيرات لدراسة التجربة، وهذه البيانات يمكن استخدامها لتحديد أولويات تلك التي تقع داخل المناطق التنظيمية المحتملة في أنواع الخلايا والأنسجة ذات الصلة. وعلاوة على ذلك، في الحالات التي يكون فيها البديل هو في ذروته في الشذرة وما يليها لبروتين معين، ويمكن لهذه البيانات توفير يؤدي المحتملة فيما يتعلق TF معين (ق) أو العوامل المشتركة التي الملزم قد تؤثر.
بعد ذلك، يتم فحص المتغيرات الناتجة الأولوية تجريبيا للتحقق من صحة توقع البروتين تعتمد على التركيب الوراثي ملزمة باستخدام EMSA 21،22. EMSA يقيس التغير في هجرة بنسبة ضئيلة على غير الحد هلام TBE. وحضنت بنسبة ضئيلة fluorescently المسمى معالمحللة النووي، وربط العوامل النووية في اعاقة حركة جزئية على هلام. في هذه الطريقة، بنسبة ضئيلة أن تربط بين العوامل النووية المزيد من سيقدم على أنه إشارة الفلورسنت أقوى على المسح الضوئي. والجدير بالذكر، EMSA لا تتطلب التنبؤات حول بروتينات معينة التي ستتأثر ملزم.
وبمجرد تحديد المتغيرات التي تقع داخل المناطق التنظيمية توقع وتكون قادرة على العوامل النووية ملزمة بشكل مختلف، واستخدام طرق الحسابية للتنبؤ TF معين (ق) الذي ملزمة لأنها قد تؤثر. ونحن نفضل استخدام CIS-BP 23،24، RegulomeDB 25، UniProbe 26، وJASPAR 27. وبمجرد تحديد مرشح TFS، هذه التنبؤات يمكن اختبار على وجه التحديد باستخدام أجسام مضادة ضد هذه TFS (EMSA-supershifts وضبا-الغرب). ينطوي على EMSA-supershift إضافة الضد-TF محددة لالمحللة النووي وبنسبة ضئيلة. ونتيجة إيجابية في EMSA-supershift هي represented تحولا آخر في الفرقة EMSA، أو خسارة من الفرقة (إعادة النظر في المرجع 28). في ضبا التكميلي، يتم تحضين على الوجهين بنسبة ضئيلة من 5'المعقدة البيروكسيديز تحتوي على البديل و 20 قاعدة الزوج المرافقة النيوكليوتيدات مع المحللة النووي من نوع من الخلايا ذات الصلة (ق) لالتقاط أي عوامل النووية ملزمة على وجه التحديد oligos. ويجمد بنسبة ضئيلة على الوجهين النووي مجمع عامل من قبل streptavidin ميكروبيدات في عمود المغناطيسي. يتم جمع العوامل النووية ملزمة مباشرة من خلال شطف 29،48. ويمكن بعد ذلك التنبؤات ملزم يتم تقييمها بواسطة لطخة غربية باستخدام الأجسام المضادة المحددة للبروتين. في الحالات التي لا توجد توقعات واضحة، أو الكثير من التوقعات، وelutions من البديل سحب هبوطا من التجارب ضبا يمكن إرسالها إلى نواة البروتينات لتحديد TFS مرشح باستخدام مطياف الكتلة، والتي يمكن بعد ذلك يمكن التحقق من صحة استخدام هذه الموصوفة سابقا أساليب.
في ما تبقى من الاعتده، يتم توفير بروتوكول مفصلة عن EMSA وضبا تحليل المتغيرات الجينية.
