Sekvensering og genotyping baserte studier, inkludert Genome-Wide Association Studies (GWAS), kandidat locus studier, og dyp-sekvense studier har identifisert mange genetiske varianter som er statistisk assosiert med en sykdom, trekk, eller fenotype. I motsetning til tidligere prediksjoner, er de fleste av disse variantene (85-93%) lokalisert i ikke-kodende regioner og endrer ikke aminosyresekvensen til proteiner 1,2. Tolking av funksjonen av disse ikke-kodende varianter og å bestemme de biologiske mekanismer som forbinder dem til den tilhørende sykdom, har egenskap, eller fenotype vist seg vanskelig 3-6. Vi har utviklet en generell strategi for å identifisere de molekylære mekanismene som lenker varianter til en viktig mellom fenotype – genuttrykk. Denne rørledningen er spesielt utviklet for å identifisere modulering av TF bindende av genetiske varianter. Denne strategien kombinerer beregnings tilnærminger og molekylærbiologiske teknikker som mål å forutsibiologiske effekter av kandidat varianter i silico og bekrefte disse spådommene empirisk (figur 1).
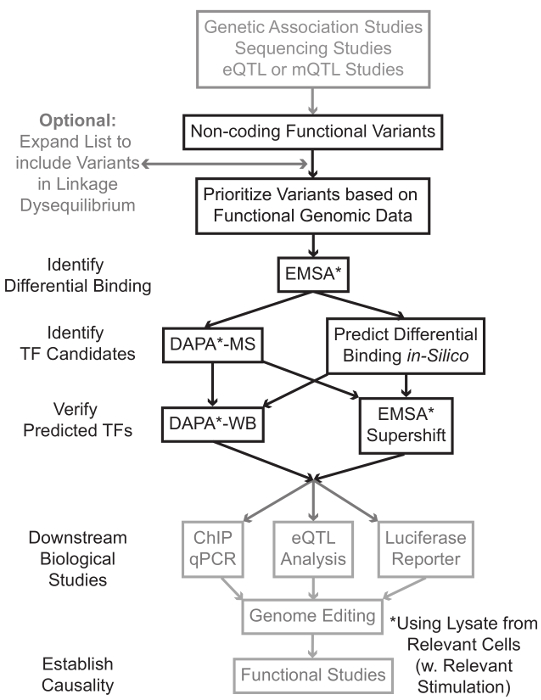
Figur 1:.. Strategisk tilnærming for analyse av ikke-kodende genetiske varianter trinn som ikke er inkludert i detaljert protokoll i forbindelse med dette manuskriptet er skyggelagt i grått Klikk her for å se en større versjon av dette tallet.
I mange tilfeller er det viktig å begynne med å utvide listen over varianter for å inkludere alle de i høy linkage-ulikevekt (LD) med hver statistisk tilhørende variant. LD er et mål på ikke-tilfeldig forening av alleler ved to forskjellige kromosomale stillinger, som kan måles ved R 2 7. R2 er et mål på linKage ulikevekt mellom to varianter, med en r 2 = 1 betegner perfekt kobling mellom to varianter. Alleler i høy LD er funnet å co-skille på kromosomet over forfedrenes populasjoner. Nåværende genotyping arrays omfatter ikke alle kjente varianter i det menneskelige genom. I stedet, de utnytte LD innenfor det menneskelige genom, og inkluderer en undergruppe av de kjente variantene som fungerer som stedfortredere for andre varianter innenfor en bestemt region av LD 8. Således kan en variant uten noe biologisk konsekvens være assosiert med en spesiell sykdom fordi det er i LD med den kausale variant-varianten med en meningsfull biologisk virkning. Prosedyremessig anbefales det å konvertere den nyeste utgaven av 1000 genomer projisere 9 variant samtalefiler (vcf) i binærfiler som er kompatible med plink 10,11, en åpen kildekode-verktøy for hele genomet forening analyse. Deretter alle andre genetiske varianter med LD r 2> 0,8 med hver inngang genetisk vaRiant kan identifiseres som kandidater. Det er viktig å bruke riktig referansegruppen for dette trinn for eksempel hvis en variant ble identifisert i fagene europeisk opphav, data fra personer av samme herkomst bør brukes for LD ekspansjon.
LD ekspansjon resulterer ofte i en rekke kandidat varianter, og det er sannsynlig at bare en liten brøkdel av disse bidrar til sykdom mekanisme. Ofte er det umulig å eksperimentelt undersøke hver av disse variantene individuelt. Det er derfor nyttig å utnytte de tusenvis av offentlig tilgjengelige funksjonell genomikk datasett som et filter for å prioritere varianter. For eksempel har KODE konsortiet 12 utført tusenvis av chip-seq eksperimenter som beskriver binding av TFS og co-faktorer, og histone karakterer i et bredt spekter av sammenhenger, sammen med kromatin tilgjengelighets data fra teknologier som DNase-seq 13, ATAC -seq 14, og FAIRE-seq 15. Databases og webservere som UCSC Genome Browser 16, Roadmap Epigenomics 17, Blueprint epigenome 18, Cistrome 19, og tilordne 20 gir gratis tilgang til data som produseres av disse og andre eksperimentelle teknikker over et bredt spekter av celletyper og vilkår. Når det er for mange varianter å undersøke eksperimentelt, kan disse dataene brukes til å prioritere de som ligger innenfor sannsynlig regulatoriske regioner i relevante celle- og vevstyper. Videre, i tilfeller der en variant er innenfor en chip-seq topp for et bestemt protein, kan disse dataene gir potensielle kunder med hensyn til spesifikke TF (e) eller co-faktorer som bindende kan påvirke.
Deretter blir de resulterende prioritert variantene vist eksperimentelt å validere spådd genotype-avhengige proteinbinding ved hjelp av EMSA 21,22. EMSA måler endringen i migreringen av oligo på en ikke-reduserende TBE gel. Fluorescensmerkede oligo inkuberes med denatom lysat, og binding av nukleære faktorer vil retardere bevegelsen av oligo på gelen. På denne måte oligo som har bundet flere nukleære faktorer vil presentere som en sterkere fluorescerende signal på skanning. Spesielt, har EMSA ikke krever spådommer om spesifikke proteiner som binding vil bli berørt.
Når varianter er identifisert som befinner seg innenfor forutsagte regulatoriske områder og er i stand til differensielt bindende nukleære faktorer, blir beregningsmetoder anvendt for å forutsi den aktuelle TF (e) hvis binding de kan påvirke. Vi foretrekker å bruke cis-BP 23,24, RegulomeDB 25, UniProbe 26, og Jaspar 27. Når kandidaten TFS er identifisert, kan disse spådommene være spesielt testet ved hjelp av antistoffer mot disse TFS (EMSA-supershifts og DAPA-Westerns). En EMSA-supershift involverer tilsetning av et TF-spesifikt antistoff til det nukleære lysatet og oligo. Et positivt resultat i en EMSA-supershift er represented som en ytterligere forskyvning i EMSA band, eller et tap av bandet (anmeldt i referanse 28). I den komplementære DAPA, er en 5'-biotinylert oligo duplex inneholder variant og 20 basepar flankerer nukleotider inkubert med atom lysat fra relevant celletype (r) for å fange opp eventuelle atom faktorer spesifikt binde oligos. Oligo duplex-nukleær faktor komplekset er immobilisert streptavidin av mikroperler i en magnetisk kolonne. De bundet kjernefysiske faktorer samles direkte gjennom eluering 29,48 -kan. Bindings forutsigelser kan deretter bli vurdert av en Western blot ved anvendelse av antistoffer som er spesifikke for proteinet. I tilfeller der det er ingen åpenbare forutsigelser, eller for mange spådommer, de elueringer fra variant pull-downs av DAPA eksperimenter kan sendes til en proteomikk kjerne for å identifisere kandidat TFS hjelp massespektrometri, som senere kan bli validert ved hjelp av disse tidligere beskrevet metoder.
I det gjenværende av article er detaljert protokoll for EMSA og DAPA analyse av genetiske varianter forutsatt.
