Секвенирования и генотипирования на основе исследований, включая геномные исследования ассоциации (GWAS), кандидат исследований локуса, и глубокие секвенирования исследования, выявили множество генетических вариантов, которые статистически связаны с заболеванием, признаком, или фенотипа. Вопреки ранних предсказаний, большинство из этих вариантов (85-93%) расположены в некодирующих областей и не изменяют аминокислотную последовательность белков 1,2. Интерпретируя функции этих некодирующих вариантов и определения биологических механизмов , связывающих их с сопутствующей болезни, признак или фенотип оказался непростым 3-6. Мы разработали общую стратегию для выявления молекулярных механизмов, связывающих варианты к важному промежуточного фенотипа – экспрессии генов. Этот трубопровод разработан специально для идентификации модуляции TF связывания генетических вариантов. Эта стратегия сочетает в себе вычислительные подходы и методы молекулярной биологии, направленных предсказатьбиологические эффекты вариантов кандидатов в силикомарганца, и проверить эти предсказания эмпирически (рисунок 1).
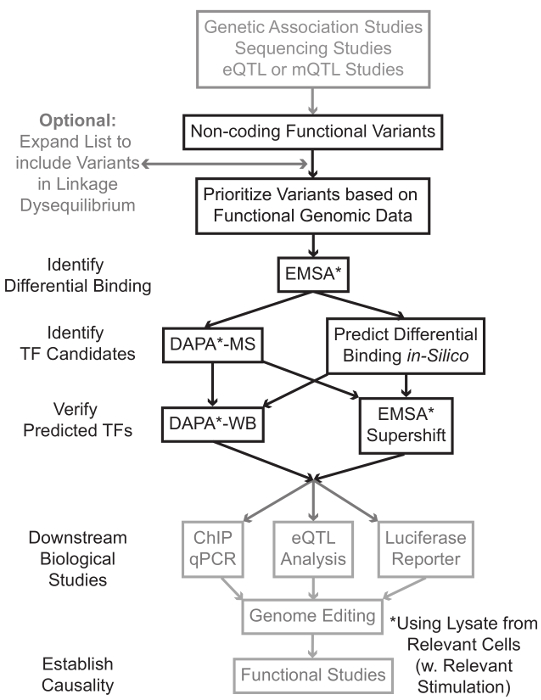
Рисунок 1:.. Стратегический подход для анализа некодирующих генетические варианты шагов, которые не включены в подробный протокол , связанный с этой рукописи заштрихованы серым цветом Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
Во многих случаях, важно, чтобы начать путем расширения списка вариантов, чтобы включить все те, в высокой рычажной-неравновесия (LD) с каждым статистически связанным вариантом. ЛД является мерой неслучайной ассоциации аллелей двух различных хромосомных положениях, которые могут быть измерены с помощью R 2 статистики 7. R 2 представляет собой меру линКейдж неравновесие между двумя вариантами, с R 2 = 1 , обозначающее совершенной связи между двумя вариантами. Аллели в высокой LD оказываются совместно разделять на хромосоме через родовые популяции. Современные генотипирования массивы не включают в себя все известные варианты в геноме человека. Вместо этого они используют ЛД внутри генома человека и включают в себя подмножество известных вариантов , которые действуют в качестве прокси для других вариантов в пределах определенной области LD 8. Таким образом, вариант без какого-либо биологического последствие может быть связан с конкретным заболеванием, потому что это в LD с причинным вариантом: вариант с значимого биологического эффекта. С процедурной точки зрения рекомендуется преобразовать последний выпуск 1000 геномов проекта 9 файлов вариант вызова (VCF) в двоичные файлы , совместимые с Plink 10,11, инструмент с открытым исходным кодом для анализа всего генома ассоциации. Впоследствии все другие генетические варианты с LD г 2> 0,8 с каждым входом генетической ваRiant могут быть идентифицированы в качестве кандидатов. Важно использовать соответствующий ссылочный населения для этого , например , Step – , если вариант был выявлен у пациентов европейского происхождения, данные от субъектов аналогичного происхождения должны быть использованы для расширения LD.
Расширение LD часто приводит десятки вариантов кандидатов, и вполне вероятно, что лишь небольшая часть из них внести свой вклад в механизм заболевания. Часто, это неосуществимо экспериментально исследовать каждый из этих вариантов по отдельности. Поэтому полезно использовать тысячи доступных функциональных публично геномных наборов данных в качестве фильтра для определения приоритетности вариантов. Например, КОДИРОВАНИЯ консорциум 12 выполнил тысячи чиповых-сл экспериментов , описывающих связывание ТФ и сопутствующих факторов и гистонов марок в широком диапазоне контекстов, наряду с данными хроматина доступности от технологий , таких как ДНКазы сл 13, ATAC -seq 14 и FAIRE-15 сл. Databтузы и веб – серверы , такие как браузера УСК генома 16, Дорожная карта Epigenomics 17 Blueprint эпигеном 18, Cistrome 19 и ReMap 20 обеспечивают свободный доступ к данным , полученных с помощью этих и других экспериментальных методов в широком диапазоне типов и условий клеток. Когда есть слишком много вариантов, чтобы проверить экспериментально, эти данные могут быть использованы для приоритеты тех, которые находятся в пределах возможных регуляторных областей в соответствующих тканей и клеток типов. Кроме того, в тех случаях, когда вариант в микросхеме-сл пика для специфического белка, эти данные могут обеспечить потенциальных клиентов, как к специфическому TF (ами) или кофакторов, связывание может затрагивающий.
Далее, полученные по приоритетам варианты экспериментально скринингу для проверки связывания с использованием EMSA 21,22 предсказанную генотип-зависимого белка. EMSA измеряет изменение миграции олиго на невосстановительной геле КЭ. Флуоресцентно меченных олиго инкубируют сядерный лизат, и связывание ядерных факторов замедлит движение олиго на геле. Таким образом, олиго-, который связан больше ядерных факторов представит в качестве более сильного флуоресцентного сигнала при сканировании. Следует отметить, что EMSA не требует предсказания о специфических белков, связывание будут затронуты.
После того, как варианты идентифицируются, которые расположены в пределах прогнозируемых регуляторных областей и способны дифференцированно связывания ядерных факторов, вычислительные методы используются для прогнозирования конкретный TF (ы), связывание которого они могут повлиять. Мы предпочитаем использовать CIS-BP 23,24, RegulomeDB 25, UNIProbe 26 и 27 Джаспер. После того, как кандидат ТФ определены, эти прогнозы могут быть специально протестированы с использованием антител против этих ТФ (EMSA-supershifts и Dapa-вестерны). EMSA-supershift включает добавление антитела к TF специфичные к ядерной лизата и олиго. Положительный результат в EMSA-supershift является магнезииesented как дальнейший сдвиг в полосе EMSA или потери полосы (обзор в ссылке 28). В дополнительном Dapa, 5'-биотинилированные олиго дуплекс, содержащий вариант и 20 пар оснований нуклеотидов фланговый инкубируют с ядерными лизата из соответствующего типа (ов) клеток, чтобы захватить любые ядерные факторы, специфически связывающих олигонуклеотидов. Фактор-комплекс олиго дуплексной ядерная скованной стрептавидином микрогранулы в магнитном колонке. Связанные ядерные факторы собираются непосредственно через элюирования 29,48. Связующие предсказания затем могут быть оценены с помощью Вестерн-блоттинга с использованием антител, специфичных к белку. В тех случаях, когда нет никаких очевидных предсказаний, или слишком много прогнозов, то элюции из вариантов отжимания экспериментов Dapa могут быть отправлены в активную зону протеомики для выявления кандидатов ТФ с использованием масс-спектрометрии, которые впоследствии могут быть проверены с помощью этих ранее описанных методы.
В оставшейся части статьи обе, подробный протокол для EMSA и Dapa анализа генетических вариантов предусмотрена.
