Figure 1 presents a flowchart of the swab sampling protocol. This protocol consists of four main steps; 1) sample collection, 2) sample storage and transportation, 3) viral RNA purification and concentration and 4) RT-qPCR assay and genotyping.
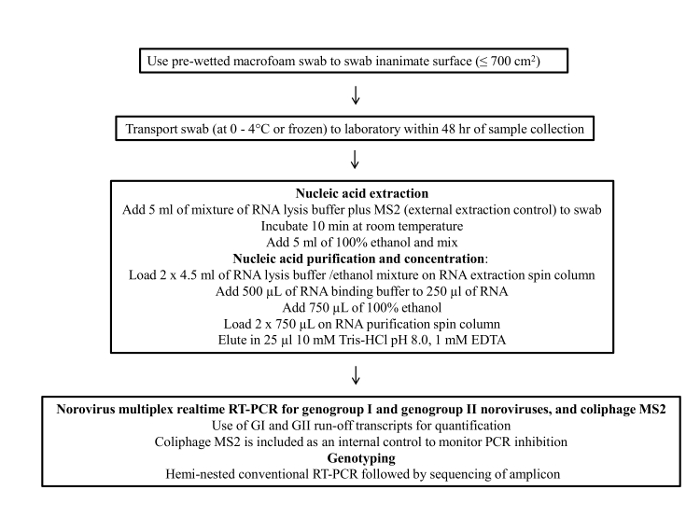
Figure 1: Flow chart of the final protocol for environmental surface sampling of norovirus Please click here to view a larger version of this figure.
Table 1 summarizes the results from 34 swab samples that were collected from a cruise ship that had reported cases of suspected norovirus gastroenteritis during a voyage. Swab samples were extracted and tested in duplicate for norovirus by multiplex real-time RT-qPCR. Seventeen (18.5%) samples tested positive including 8 samples from surfaces in cabins where passengers with norovirus symptoms had stayed and 9 from common areas on the ship. The number of genomic copies per sample were calculated from the Ct values (which ranged from 16 to 31) using a standard curve of a norovirus GII.7 RNA transcript. The median number of genome copies from swab samples in cabins was 3.6 log10(range: 2.4 – 4.5 log10 RNA copies), which was significantly higher than the genome copies from common areas (range: 1.2 – 2.1 log10 genome copies) (P <0.001). Four (23.5%) of the 17 positive swab samples were able to be genotyped, and all samples had identical GII.1 sequences.
| Location environmental swap collecteda | Sample Point Description | Average Ct value (# positive of the total number of samples tested) | Genotype | Norovirus RNA copy number per sampled areac |
| Atrium | Handrails | 34.3 (1/2) | GII | 16 |
| Cabin A | Toilet seat | 31.4 (2/2) | GII.b | 31,217 |
| Cabin A | Faucet | 37.5 (1/2) | GII | 491 |
| Cabin A | Door handle | 35.0 (2/2) | GII | 2,675 |
| Cabin A | Remote control | 38.6 (2/2) | GII.1b | 233 |
| Cabin B | Toilet seat | 33.5 (2/2) | GII.1b | 986 |
| Lido | Ice Cream Machine | 34.2 (2/2) | GII | 16 |
| Lido | Table Condiments | 35.2 (1/2) | GII | 15 |
| Lido | Table Top | 35.3 (1/2) | GII | 14 |
| Pizzeria | Counter surface | 35.7 (1/2) | GII | 14 |
| Main Galley | Fun-time Machine | 37.1(1/2) | GII | 64 |
| Vending Machine | Touchable Surfaces | 38.8 (1/2) | GII | 18 |
| Crew lounge | Keyboard Surface and Mouse | 36.8 (1/2) | GII | 80 |
| Cabin C | Faucet and door handle | 31.6 (2/2) | GII.1b | 26,458 |
| Cabin C | Telephone | 36.4 (2/2) | GII | 1,035 |
| Cabin C | Keyboard | 33.0 (2/2) | GII | 1,317 |
| Medical center | Clipboard | 36.0 (2/2) | GII | 113 |
| aCabin A, B and C has been occupied by individuals who had been clinically ill with viral gastoenteristis symptoms. | ||||
| bFour of the 17 GII-positive swab samples could be genotyped. | ||||
| cRNA copies were caluated based on a standard curve of norovirus GII.7 RNA transcripts. | ||||
| Note: above date were slightly modified from the original article6. | ||||
Table 1: Results from 34 swab samples that were collected from a cruise ship that had reported cases of suspected norovirus gastroenteritis during a voyage.
Figure 2 shows the relationship of Ct values determined by RT-qPCR and the ability to sequence these samples. In total, 127 out of 217 swab samples tested positive for GII norovirus by RT-qPCR. The samples displayed a wide range (12-40) of Ct values. In total, 29 (22.8%) of the RT-qPCR positive samples could be genotyped. Swab samples with Ct values below 27 and ranging between 28-31 were genotyped at rates of 100% and 72.2%, respectively. In contrast, only 22.0% and 1.6% of swab samples with Ct values of 32-36 and 37-40, respectively, produced hemi-nested amplicons that could be sequenced successfully.
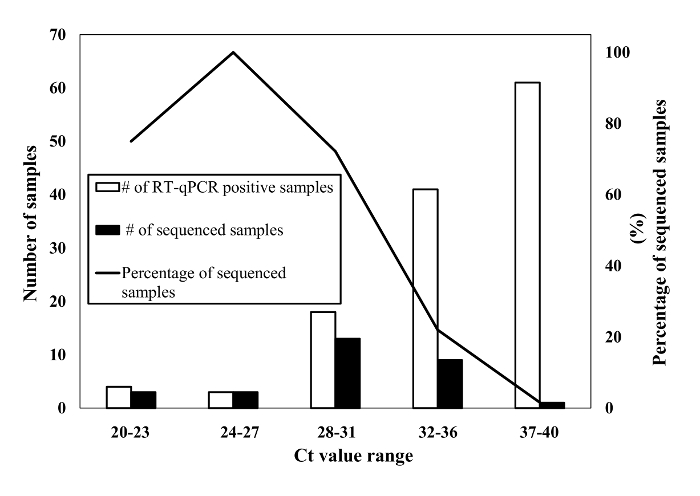
Figure 2: Results of RT-qPCR screening and sequencing of swab samples that had been collected during confirmed norovirus outbreaks. White bars represent the number of RT-qPCR positive swab samples virus. Human norovirus nucleic acids in those RT-qPCR positive samples were amplified using hemi-nested PCR assay and then sequenced for confirmation of genotyping. Black bars and line represent the number and the percentage of genotype confirmed swab samples, respectively. Please click here to view a larger version of this figure.
| Ct value range | Number of RT-qPCR positive samples | Number (%) of sequence confirmed samples a |
| 20-23 | 4 | 3 (75) |
| 24-27 | 3 | 3 (100) |
| 28-31 | 18 | 13 (72.2) |
| 32-36 | 41 | 9 (22.0) |
| 37-40 | 61 | 1 (1.6) |
| a) hemi nested PCR | ||
Table 2: RT-qPCR and genotyping results of swab samples.
| Sample Number | LOCATION | Description of surface | Surface area (cm2) | Date/time of sample collection | Clean (Y/N) | If cleaned, what disinfectant was used? | Detailed description of surface material and location |
| 1 | Room# 7-1302 | Toilet Seat | 700 cm2 | 6/26/2016; 9:00 am | No | not applicable | Toilet seat [top surfaces), and hard plastoc |
| 2 | Room #7-1330 | Telephone handle | 500 cm2 | 6/26/2016; 9:25 am | Yes | 1,000 ppm bleach | Hard plastic, rubber button |
Supplementary Table 1: Example of sample description form.
| genogroup / virus | Name oligonucleotide primer/probe | Sequence 5΄→3΄ | Reference |
| GI | Cog1F | CGY TGG ATG CGI TTY CAT GA | 17 |
| Cog1R | CTT AGA CGC CAT CAT CAT TYA C | 17 | |
| G1SKF | CTG CCC GAA TTY GTA AAT GA | 17 | |
| G1SKR | CCA ACC CAR CCA TTR TAC A | 17 | |
| Ring1E-probe | FAM – TGG ACA GGR GAY CGC – MGBNFQa | This study | |
| GII | Cog2F | CAR GAR BCN ATG TTY AGR TGG ATG AG | 17 |
| Cog2R | TCG ACG CCA TCT TCA TTC ACA | 17 | |
| Ring2-primer | TGG GAG GGC GAT CGC AAT CT | 17 | |
| G2SKR | CCR CCN GCA TRH CCR TTR TAC AT | 17 | |
| Ring2-probe | Cy5 or QUASAR 670 – TGG GAG GGC GAT CGC AAT CT – BHQ2b | 17 | |
| MS2 | MS2F | TGG CAC TAC CCC TCT CCG TAT TCA CG | 18 |
| MS2R | GTA CGG GCG ACC CCA CGA TGA C | 18 | |
| MS2P-probe | HEX – CAC ATC GAT AGA TCA AGG TGC CTA CAAGC – BHQ1c | 18 | |
| aGI TaqMan probe is 5’-labeled with 6-carboxyfluorescein (FAM) and 3’-labeled with MGBNFQ (Minor-groove Binding site) | |||
| bGII TaqMan probe is 5’-labeled with Cy5 or Quasar 670 and 3’-labeled with Black Hole quencher; Black Hole Quencher (BHQ) 2 used due to availability, BHQ 3 is preferred. | |||
| cMS2 TaqMan probe is 5’-labeled with HEX and 5’-labeled with BHQ1 | |||
Supplementary Table 2: Oligonucleotide primers and probes information.
| Component | Volume per reaction (µl) | Final concentration |
| 2x RT-PCR Buffer* | 12.5 | 1x |
| Nuclease-free water* | 1.08 | n/a |
| Detection Enhancer* | 1.67 | n/a |
| Cog 1F (10 µM) | 1 | 400 nM |
| Cog 1R (10 µM) | 1 | 400 nM |
| Ring 1E-probe (10 µM) | 0.5 | 200 nM |
| Cog 2F (10 µM) | 1 | 400 nM |
| Cog 2R (10 µM) | 1 | 400 nM |
| Ring 2-probe (10 µM) | 0.5 | 200 nM |
| MS2F (10 µM) | 0.25 | 100 nM |
| MS2R (10 µM) | 0.25 | 100 nM |
| MS2P-probe (10 µM) | 0.25 | 100 nM |
| 2x RT-PCR enzyme * | 1 | 1x |
| Master Mix volume | 22 | |
| * included in the real time RT-PCR kit | ||
Supplementary Table 3: Master mix for multiplex real-time GI/GII/MS2 norovirus RT-PCR
| Type of control | Result interpretation | ||
| Sample | Negative control | Should be negative | |
| Positive control | Should be positive | ||
| Negative sample | Sample is negative if each Ct value is undetectable for GI/GII | ||
| Positive sample | Ct values (GI/GII) of both replicates is < 38; this (arbitrarily) cut-off needs to be determined experimentally for each realtime PCR platform and kit | ||
| Tentative positive sample | Sample is tentatively positive if Ct value (GI/GII) of one replicate is <38 | ||
| Internal process control (MS2) | Samples with a threshold cycle (Ct) value of ≥32 for MS2 should be retested undiluted and 1/10 diluted. | ||
| Parameter | Acceptable value | ||
| Standard curve using norovirus RNA transcripts | R2 | >0.97 | |
| Efficiency | 90% to 115% | ||
Supplementary Table 4: Controls to include in each RT-qPCR test for detection of norovirus in environmental swab samples.
| Component | Final concentration | vol/rxn (µl) |
| 5x RT-PCR buffer | 1x | 5.00 |
| dNTP mix (10 mM) | 0.4 mM | 1.00 |
| Enzyme mix (RT and Taq) | 1.00 | |
| Forward primera | 0.5 µM | 0.50 |
| Reverse primer a | 0.5 µM | 0.50 |
| Rnase inhibitor (20 U/µl) | 20 U | 1.00 |
| Rnase-free water | 11.00 | |
| Total Volume | 20.00 | |
| aforward and reverse primer sets for GI and GII group are Cog1F + G1SKR, and Cog2F + G2SKR, respectively. | ||
| Second round RT-PCR | ||
| Component | Final concentration | vol/rxn (µl) |
| 5x RT-PCR buffer | 1x | 5.00 |
| dNTP mix (10 mM) | 0.4 mM | 1.00 |
| Enzyme mix (RT and Taq) | 1.00 | |
| Forward primerb | 0.5 µM | 0.50 |
| Reverse primerb | 0.5 µM | 0.50 |
| Rnase inhibitor (20 U/µl) | 20 U | 1.00 |
| Rnase-free water | 14.00 | |
| Total Volume | 23.0 | |
| bforward and reverse primer sets for GI and GII group are G1SKF + G1SKR, and Ring2 primer + G2SKR, respectively. | ||
| Note: above informaion was slightly modified from the original article19 | ||
Supplementary Table 5: Hemi-nested RT-PCR Master mix.
| Phase I | Field Sampling | 1. Check macrofoam swab kits (e.g., expiration date, tube leakages, and swab wetness) |
| 2. Swab surface (limit surface area to ≤100 inch2 (645 cm2)) | ||
| 3. Place swabs into the transport tubes, and tighten the caps securely to prevent leaking during shipping | ||
| 4. Place swab kit in a ziplock-bag | ||
| Phase II | Sample transport and storage | 1. Sampled swabs should be stored -70 °C (or -20 °C) |
| 2. Transporting swab samples to laboratory at 0-4 °C (cold-packs) in an insulated container and ship within 48 hr of collecting swabs | ||
| Phase III | RNA etraction | 1. Check lysis buffer (e.g., expiration data) |
| 2. Add MS2 as an internal control into lysis buffer before adding 100% ethanol | ||
| RNA purification and concentration | 3. Clean lab bench and small equipment using RNA RNase removal solution | |
| 4. Change gloves frequently during steps to avoid a RNA cross-contamination | ||
| Phase IV | RT-PCR Assay (QA/QC) | 1. Include quantified norovirus transcript RNAs (GI and GII) for each RT-qPCR assay to control for variations in Ct values for each PCR run |
| 2. Use absence of MS2 signal to monitor presence of PCR inhibitors |
Supplementary Table 6: Checklist for CDC's environmental sampling procedure.
