Previously, we have shown that genetic deletion of Bcl11a by in utero electroporation impairs radial migration of late-born upper-layer projection neurons10. Electroporation of a DNA plasmid vector containing Cre-IRES-GFP efficiently deleted Bcl11a in conditional Bcl11aflox/flox brains11. When we analyzed E14.5 electroporated brains three days after the electroporation, most control neurons had migrated into the CP, whereas many Bcl11a mutant neurons were stalled along the migratory route in the IZ and VZ/SVZ. Moreover, we found an increase in the number of multipolar cells at the expense of bipolar cells specifically in the IZ upon Bcl11a deletion suggesting defects in polarization10.
To directly and dynamically analyze migratory behavior of Bcl11a mutant neurons we used time-lapse confocal imaging of migrating neurons in organotypic slice culture prepared from electroporated brains. Overview time-lapse series using a 10X objective with a numerical aperture of 0.4 confirmed our results, that Bcl11a mutant projection neurons have defects in radial migration. Within 36 h many control neurons had migrated into the CP, whereas only few Bcl11a mutant neurons had left the IZ and migrated into the CP. Interestingly, it seemed that many Bcl11a mutant neurons did not migrate directly towards the pial surface of the brain, but slowed down migration and turned tangentially or even back towards the ventricle (Movie 1). To further analyze these findings we generated time-lapse series at a higher magnification using a 40x objective with a numerical aperture of 0.6. Our data show that Bcl11a mutant neurons failed to efficiently polarize and to continue radially oriented migration into the CP. Within 12 h, many Bcl11a mutant neurons failed to switch from a multipolar to a bipolar morphology and instead projected and retracted many processes into the surrounding environment (Movie 2, Figure 1A).
Furthermore, we traced the migration paths of individual neurons in different time-lapse series and used these to calculate specific parameters of migrating neurons. We found that Bcl11a mutant neurons repetitively undergo phases of several hour duration with reduced migration speed and random-like orientation changes (Movie 3; Figure 1B, red arrowheads). In particular, the speed profile of Bcl11a mutant neurons was shifted from higher speed (25 µm/h) towards lower speed (5 µm/h) in comparison to control neurons (Figure 1C). In line with this, the overall migration speed of Bcl11a mutant neurons was significantly reduced from 10.34 ± 0.34 µm/h in controls to 6 ± 0.54 µm/h (p = 2.4889E-05) (Figure 1D). Finally, the deviation from directed migration towards the pial surface of the brain was significantly increased from 17.15 ± 2.13° in control to 40.16 ± 4.42° in Bcl11a mutant neurons (p = 0.0002) (Figure 1E). Together these results demonstrate that time-lapse confocal imaging of GFP electroporated neurons in organotypic slice culture is a valuable method to study molecular mechanisms controlling the process of neuronal migration.
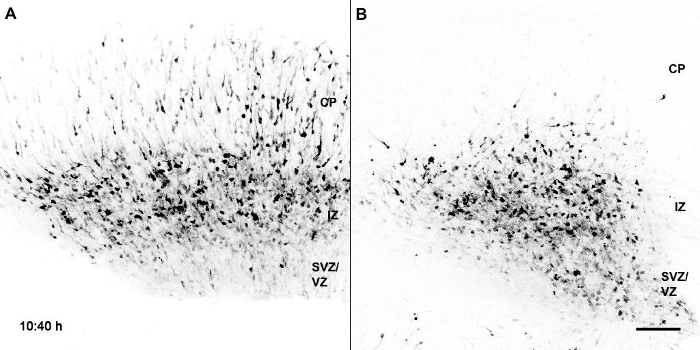
Movie 1: Migration of GFP Labeled Control in Comparison to Bcl11a Mutant Neurons in Cortical Slices. Bcl11aflox/flox brains were electroporated at E14.5 with a plasmid vector containing IRES-GFP (A) or Cre-IRES-GFP (B) and slice cultures were prepared at E16.5. VZ/SVZ; ventricular/subventricular zones; IZ, intermediate zone; CP, cortical plate. Scale bar = 100 µm. The movie is comprised of 108 frames at the rate of 5 frames/s. Reprinted from Wiegreffe et al.10 with permission from Elsevier. Please click here to view this video. (Right-click to download.)
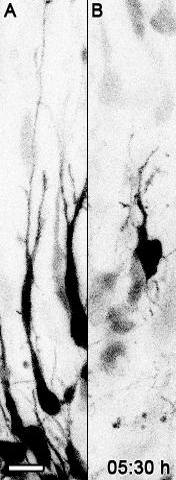
Movie 2: Polarization of a GFP Labeled Control in Comparison to Bcl11a Mutant Neurons in Cortical Slices. Bcl11aflox/flox brains were electroporated at E14.5 with a plasmid vector containing IRES-GFP (A) or Cre-IRES-GFP (B) and slice cultures were prepared at E16.5. Scale bar = 10 µm. The movie is comprised of 25 frames at the rate of 5 frames/s. Reprinted from Wiegreffe et al.10 with permission from Elsevier. Please click here to view this video. (Right-click to download.)
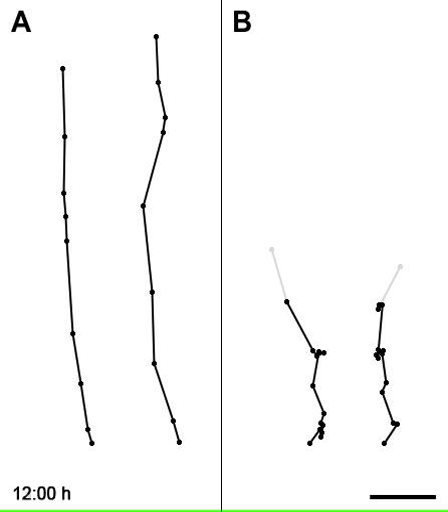
Movie 3: Animated Traces with 1 h Interval Resolution of Representative Control and Bcl11a Mutant Neurons. Traces of migrating neurons were obtained from time-lapse series of E16.5 slice cultures of Bcl11aflox/flox brains that were electroporated at E14.5 with a plasmid vector containing IRES-GFP (A) or Cre-IRES-GFP (B). Scale bar = 20 µm. Reprinted from Wiegreffe et al.10 with permission from Elsevier. Please click here to view this video. (Right-click to download.)
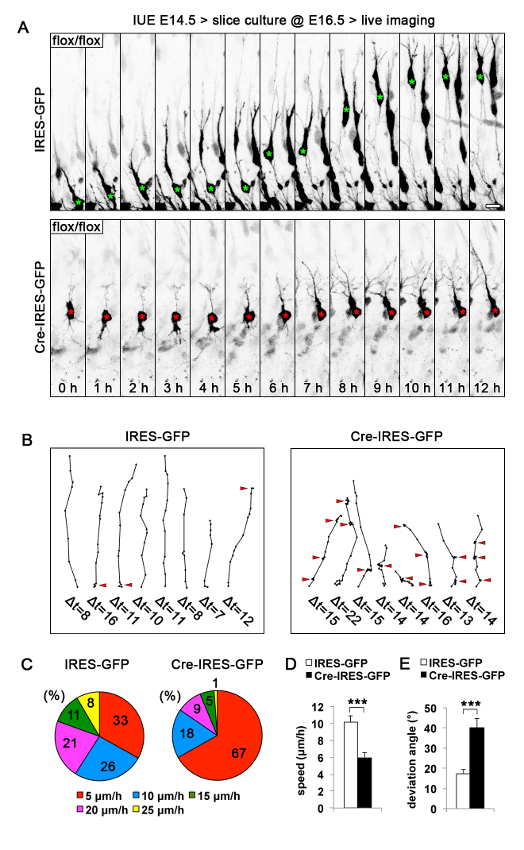
Figure 1: Migration Behavior of Bcl11a Mutant Upper-Layer Cortical Neurons.
(A) Representative images of migrating GFP positive neurons in E16.5 slice cultures from Bcl11aflox/flox brains electroporated at E14.5 with either Cre-IRES-GFP or a control vector over a total imaging period of 12 h. (B) Representative traces with 1 hour interval resolution of migrating GFP positive neurons in E16.5 slice cultures from Bcl11aflox/flox brains electroporated at E14.5 with Cre-IRES-GFP or a control vector over total imaging periods of up to 22 h. Bcl11a mutant neurons frequently undergo repetitive phases of reduced migration speed and randomly changed orientation (marked by red arrowheads). (C) Speed profiles calculated from traces of migrating neurons as shown in B. (D-E) Quantification of speed (D) and deviation angle from radial orientation (E) of migrating GFP positive neurons in E16.5 slice cultures from Bcl11aflox/flox brains electroporated at E14.5 with Cre-IRES-GFP or a control vector (n = 15). Mean ± s.e.m.; Student's t test; ***p <0.001. Scale bar = 10 μm. Reprinted from Wiegreffe et al.10 with permission from Elsevier. Please click here to view a larger version of this figure.
