DNA Polymerase Activity Assay Using Near-infrared Fluorescent Labeled DNA Visualized by Acrylamide Gel Electrophoresis
Summary
This protocol describes the characterization of DNA polymerase synthesis of modified DNA through observation of changes to near-infrared fluorescently labeled DNA using gel electrophoresis and gel imaging. Acrylamide gels are used for high resolution imaging of the separation of short nucleic acids, which migrate at different rates depending on size.
Abstract
For any enzyme, robust, quantitative methods are required for characterization of both native and engineered enzymes. For DNA polymerases, DNA synthesis can be characterized using an in vitro DNA synthesis assay followed by polyacrylamide gel electrophoresis. The goal of this assay is to quantify synthesis of both natural DNA and modified DNA (M-DNA). These approaches are particularly useful for resolving oligonucleotides with single nucleotide resolution, enabling observation of individual steps during enzymatic oligonucleotide synthesis. These methods have been applied to the evaluation of an array of biochemical and biophysical properties such as the measurement of steady-state rate constants of individual steps of DNA synthesis, the error rate of DNA synthesis, and DNA binding affinity. By using modified components including, but not limited to, modified nucleoside triphosphates (NTP), M-DNA, and/or mutant DNA polymerases, the relative utility of substrate-DNA polymerase pairs can be effectively evaluated. Here, we detail the assay itself, including the changes that must be made to accommodate nontraditional primer DNA labeling strategies such as near-infrared fluorescently labeled DNA. Additionally, we have detailed crucial technical steps for acrylamide gel pouring and running, which can often be technically challenging.
Introduction
DNA polymerases perform accurate and efficient DNA synthesis and are essential to maintaining genome integrity. The ability to synthesize hundreds of nucleotides per second without making errors also makes DNA polymerases essential tools in molecular biology and biotechnology. However, these properties also limit the applications for M-DNA substrates; generally speaking, natural DNA polymerases cannot synthesize many potentially valuable M-DNA substrates, likely due to the high selective pressure against using non-standard substrates in vivo. Many groups have developed directed evolution approaches to generate mutant DNA polymerases capable of M-DNA synthesis1a,2,3,4,5; these efforts have expanded the biotechnological utility of DNA6,7,8.
To evaluate the ability of mutant DNA polymerases to synthesize M-DNA, we9,10, and others11,12,13 typically use in vitro measurements of DNA polymerase activity, which are described in this manuscript. In these experiments, DNA polymerases are co-incubated with a labeled primer/template duplex and nucleoside triphosphate substrates; the products are evaluated by gel electrophoresis. Depending on the specific experimental question, mutant DNA polymerases, modified primers, modified templates, or modified nucleoside triphosphates can be used, enabling the systematic biochemical evaluation of the mutant enzyme activity.
Historically, these assays have relied on a 5' radioactive label to track DNA synthesis; most commonly, 32P and 33P have been used; typically, labeling is achieved using T4 polynucleotide kinase11. However, due to the finite lifetime and relatively high cost of radioactive labels and their safe disposal, our group instead uses a synthetic 5' near-infrared fluorophore labeled DNA. Using a relatively low cost near-infrared gel imager, we have observed similar detection limits to prior studies using radioactive labels (unpublished results). We have successfully reproduced past observations9, and we have not observed any large quantitative difference with previously measured rate constants (unpublished results).
To analyze the size of DNA, and thus, the extent of DNA synthesis, we rely on polyacrylamide gel electrophoresis methods developed originally for Sanger sequencing14 before the advent of capillary electrophoresis15. The distance of separation or mobility can be used as a measurement of molecular weight; large format, vertical polyacrylamide gels can achieve single nucleotide resolution, enabling quantitative observation of DNA oligonucleotides of varying lengths.
Collectively, these experiments are a robust method for polymerase characterization. Due to the time sensitive nature of the reactions, preparation and care is necessary to achieve reproducible results. Further, while the acrylamide gel is a highly effective way to measure DNA synthesis, as well as numerous other DNA modifying reactions, with single nucleotide resolution, it can be technically challenging. The protocol here will hopefully enable users to perform these experiments while avoiding the most common mistakes.
Protocol
1. Activity Assay
NOTE: There are two typical types of assays that are often run to characterize DNA polymerases using the methods described here. They differ in whether they qualitatively characterize overall synthesis (encompassing many steps of DNA synthesis) or whether they quantitatively focus on individual steps. We describe the necessary steps for each of these below.
NOTE: Because the assembly of materials are relatively complex, for time sensitive experiments, we recommend that all materials are assembled beforehand. The recipes of all critical components of the assay are listed below. Commercial suppliers for components are listed in the Table of Materials.
- 10x SF Buffer recipe (10x SFB)
NOTE: In our assays, we typically use the N-terminal truncation of the DNA polymerase I of Thermus aquaticus, commonly known as Stoffel fragment (SF)16,17. The recipe here is the preferred buffer for SF3,13. Other buffers may be substituted depending on the specific DNA polymerase.
NOTE: The final concentration of the 10x SF buffer components are 500 mM Tris pH 8.5, 65 mM MgCl2, 0.5 mg/mL BSA, 500 mM KCl (s), and ultrapure water. Measurements are for 1 mL of SF buffer, which is sufficient for 100-200 assays, depending on the scale. The buffer can be stored indefinitely at -20 °C.- Combine 0.5 mL of 1 M Tris, 65 µL of 1 M MgCl2, 50 µL of 10 mg/mL BSA, and 0.0373 g of KCl (s) in a 1.5 mL tube. Add 385 µL of ultrapure water to reach a final volume of 1 mL.
- 1x Storage Buffer recipe (1x SB)
NOTE: The final concentration of the 1x buffer components are 50 mM Tris pH 7.5, 1 mM DTT, 0.6 mM EDTA, and 50% glycerol. The recipe makes 20 mL of 1x SB.- Combine 0.012 g DTT, 100 µL of 0.5 M EDTA, and fill a 50 mL conical tube with 40 mL of 100 mM Tris (pH 7.5) to make 2xSB. Mix by vortexing.
- Transfer 10 mL of 2x SB to a new 50 mL conical tube and dilute with glycerol by adding 10 mL glycerol to the new conical.
- Quenching buffer orange (QBO) recipe
NOTE: When radioactive labels are used, users will often employ bromophenol blue and/or xylene cyanol as a tracking dye for electrophoresis progress. However, these dyes will weakly fluoresce in the near-infrared region, interfering with the DNA signal. Thus, we use Orange G in their place for all reactions that contain samples. While we have found Orange G to be suitable for tracking gel loading, we have found Orange G to be less consistent as a dye that tracks electrophoresis progress; thus, we run empty lanes containing bromophenol blue on the external lanes of the gel that do not contain sample in order to track the gel progress.- Combine 950 µL of 95% formamide with 25 µL of 0.5 M EDTA, and 25 µL ultrapure water. Add 1 scoop of orange G dye (~10 mg). Mix by vortexing.
CAUTION: Formamide is toxic; wear personal protective equipment (PPE) such as gloves, eyeglasses and lab coat, and dispose as hazardous waste.
- Combine 950 µL of 95% formamide with 25 µL of 0.5 M EDTA, and 25 µL ultrapure water. Add 1 scoop of orange G dye (~10 mg). Mix by vortexing.
2. Assay Run
- Qualitative characterization of overall activity
NOTE: For this assay, we typically maintain constant concentration of M-NTPs and vary time. The goal is to evaluate the overall characteristics of the protein rather than to measure any individual step. To prepare one reaction, an equal volume of two separate components, the duplex mix (described in step 2.1.1) and the dNTP mix (described in step 2.1.2), will be prepared separately, and then combined to give the final volume of 49 µL. The addition of 1 µL 50x enzyme will then initiate the reaction. Variable time points can be taken; typically, we quench at 0, 5, 15, and 60 min.- Preparation of DNA duplex mix
NOTE: The duplex mix is composed of 10x SF buffer, primer oligonucleotide, template oligonucleotide, and ultrapure water. Total volume added to each reaction is 25 µL. 1 µL of enzyme will be added to the master mix immediately preceding the initiation of the experiment.
NOTE: For our assays, we typically use a 45-mer template and an 18-mer or 23-mer primer. While a number of different lengths can be used, we tend to use this length because the products (ranging from 18 nucleotides to 45 nucleotides) are easily resolved on a single nucleotide level using the described gel electrophoresis protocols. As oligonucleotide products increase in length, it may be challenging to resolve the products at single nucleotide resolution.
NOTE: In our experiments, we use a 5' near-infrared fluorescent dye label on the primer strand to observe the primer products. We purchase custom synthesized oligonucleotides bearing this modification; however, this label can be incorporated using any number of post-synthetic labeling strategies. The template oligonucleotide is not labeled and so it is not observed using the gel imager. For both oligonucleotides, we store the oligonucleotides at 100 µM in 10 mM Tris (pH 8), 1 mM EDTA.
NOTE: Because we are observing only the primer, a two-fold excess of template is used to ensure that all primers (the DNA species that is being observed) are annealed to the template. The final duplex concentration in this reaction is 40 nM.
NOTE: Near-infrared fluorescent dyes are often somewhat light sensitive; when possible, we cover the primer (and any solution containing the primer) with foil. This includes the polyacrylamide gel while it is running.
NOTE: Below is a representative example of synthesis of 2'F M-DNA.- Dilute primer 1 and template 1 (see Table of Materials for sequence) to 1 µM each in ultrapure water.
- For each assay reaction, in separate tubes, combine 5 µL of 10x SF buffer, 2 µL of 1 µM primer 1, 4 µL of 1 µM template 1, and 13 µL of ultrapure water in tubes compatible with a thermal cycler.
- Anneal the duplex in a thermocycler using the following program: 98 °C for 2 min, 70 °C for 5 min, 50 °C for 5 min, 40 °C for 5 min, hold at 25 °C. The specific program may vary depending on the annealing temperature of the primer/template duplex. Typically, we prefer to include at least one 5 min step at the annealing temperature, and then another 5-min step at a temperature 5 – 10 degrees below the annealing temperature.
- Preparation of 2x dNTP mixture
NOTE: Depending on the experiment, it is possible to use variable dNTPs and concentrations of dNTPs. Typically, we use 50 – 200 µM in the reaction.- Prepare a 25 µL solution containing 100 µM of each 2'F-NTP. Store the M-NTPs at 100 mM.
- Preparation of 50x protein dilution
NOTE: For long term storage, enzymes should be kept at -20 °C in 1xSB. When removed from the -20 °C freezer, enzymes should be kept on ice at all times prior to addition to the master mix. Enzyme dilutions should be made fresh from the concentrated stock each day. The concentrated enzyme stock should be returned to the -20 °C freezer immediately after use.
NOTE: Because we primarily evaluate mutant DNA polymerases, we only use enzymes that have been expressed and purified in our laboratory using established methods9,10. For commercially purchased polymerases, users should be wary of optimal buffers for different proteins and their compatibility with the buffers described herein.
NOTE: Enzyme concentrations will vary for each experiment. Here, we show a representative example for 2'F-DNA synthesis.- To make a 10 nM enzyme solution, dilute 1 µL of enzyme into 1x SB to make a 50x final enzyme concentration. The concentration of the 50x solution should be 500 nM. For example, if the stock enzyme concentration is 50 µM, add 1 µL stock enzyme to 99 µL 1x SB.
NOTE: Since the enzyme is typically stored in 50% glycerol, the solution is fairly viscous. Even if using highly accurate pipettes, we do not recommend pipetting less than 0.5 µL, considering the importance of accuracy and precision during enzyme dilution.
- To make a 10 nM enzyme solution, dilute 1 µL of enzyme into 1x SB to make a 50x final enzyme concentration. The concentration of the 50x solution should be 500 nM. For example, if the stock enzyme concentration is 50 µM, add 1 µL stock enzyme to 99 µL 1x SB.
- Initiation of assay
- Set the temperature of a dry bath to 50 °C.
- Add 24 µL of the annealed duplex mix (see step 2.1.1) to 25 µL of 2x dNTPs (see step 2.1.2).
- Remove 9.8 µL of the mixture in step 2.1.4.2 and add to 20 µL of QBO (see section 1.3 for recipe). This aliquot serves as a "no enzyme" control and should be obtained for each run.
- Add 0.8 µL of 50x enzyme (see step 2.1.3.1) to the duplex/dNTP mix. Pipette up and down with a large volume micropipette to thoroughly mix.
- After 5, 15, and 60 min, quench the reaction by removing 10 µL of each reaction solution and adding to a microcentrifuge tube containing 20 µL QBO (described in section 1.3).
- Once quenched, reactions can be stored under foil indefinitely at 4 °C or -20 °C.
NOTE: For quantitative characterization of individual steps of M-DNA synthesis, a very similar assay can be run to quantitatively obtain Michaelis-Menten parameters. This assay uses all of the same materials. Michaelis-Menten parameters can be measured for the dNTP; in this case, the most significant difference is that, rather than adding the duplex mix to dNTP mix, it adds the duplex mix with enzyme to a series of 2x dNTP solutions. Similarly, Michaelis-Menten parameters can be measured for DNA duplex. Specific conditions must be met to satisfy the assumptions needed to use the Michaelis-Menten equation. These conditions, and the basic mechanics of the assay, are well explained in a previous methods article11.
- Preparation of DNA duplex mix
3. Gel Electrophoresis
NOTE: Because pouring the gel is time sensitive, all materials are assembled beforehand. The recipes of all critical components of the assay are listed below; suppliers are listed in the Table of Materials.
- Preparing acrylamide for 20% acrylamide gel.
NOTE: This recipe makes 1 L of acrylamide mixture; approximately 120 mL of this solution is used per gel. Store this solution at room temperature under foil for up to one year.
CAUTION: Polyacrylamide is neurotoxic. It is important to wear gloves and a lab coat during all steps of gel pouring. If polyacrylamide gets on gloves, replace the gloves immediately. If acrylamide is not polymerized, place the contaminated materials in a labeled polyacrylamide waste bag.
NOTE: Premixed 40% acrylamide containing 38.67% acrylamide and 1.33% bis-acrylamide for a monomer to cross-linker ratio of 29:1 was used in this protocol.- Weigh 17 g of premixed TBE powder. Weigh six separate portions of 70 g of urea (total of 420 g).
NOTE: The urea must be added in portions. We find it best to split it into six portions. - Set up a 2 L plastic beaker on a stir plate. Add a single large stirring rod to the beaker. Cover the beaker with foil when mixing to prevent splashing.
- Add 500 mL of 40% acrylamide solution into beaker. Make sure the solution is stirring.
- Add previously weighed-out TBE. Add one aliquot of urea. Allow solution to mix. Urea dissolves slowly and may appear hazy and white upon initial addition. Slowly add the remaining aliquots of urea into the mixture over the next hour. Let the solution mix until it is completely clear and colorless.
NOTE: Typically, allow this to stir overnight. If the solution is still not dissolved, add small aliquots of water and wait for the urea to dissolve. - The addition of urea will increase the volume close to 1 L. Add water to increase the volume to exactly 1 L.
- Store in a 1 L tinted glass bottle or cover the glass bottle with aluminum foil.
- Weigh 17 g of premixed TBE powder. Weigh six separate portions of 70 g of urea (total of 420 g).
- Pouring of acrylamide gel.
NOTE: When handling glass plates, be careful to avoid contact between the glass plates and any hard surface. This is particularly important in that rough handling may cause small cracks in the glass; these cracks may not be visible until the gel is running, which occurs at a higher temperature. We use cork rings to prevent this unwanted contact.- Clean plates
- Acquire two 33 cm x 42 cm gel plates, one notched and one unnotched plate. Place plates on separate cork rings.
- Rinse plates thoroughly with soap and water. Make sure there are no soap streaks or gel spots left behind.
- Make note of which side of the plate beads less water. This side is the gel facing side.
NOTE: If the gel is run on the other side of the plate, it may make the gel transfer challenging.
- Dry plates
- Use paper towels to dry both faces of each plate.
- Use delicate task wipers to dry remaining areas. Pay special attention to the edges of plates where tape will be applied.
- Rinse the glass plates with ~70% ethanol, and wipe with task wipers.
- Make sure there are no paper towel fibers or water droplets. These can cause bubbles when pouring the gel.
- Add spacers
- Acquire 0.75 mm spacers. Run gloved fingers under DI water and gently wet the spacers with gloved fingers.
- Place spacers along the long edges of the notched plate. Clean up any water that is splashed on the rest of the plates. Make sure spacers are aligned with the glass plate and not hanging over the edge.
- Seal gel
- Put on dry gloves. Dry the edges of the glass again with delicate task wipers.
- Align the unnotched plate top above the notched plate, and slowly lower down to press the plates against each other. Check again to be sure that the edges of all plates are aligned.
- Using gel tape, place the tape across all the edges except for the top. Make sure tape is aligned evenly and sealed tightly. Tape can be applied in one motion, or each side can be done individually. Cut the ends of the tape with a razor.
- Add an additional layer of tape to the bottom of the gel. The tighter the tape, the less likely the gel will leak when pouring.
- Clip the sides of the glass sandwich with the white clamps. Add 3 clips to each side, and 4 clips to the bottom.
- Pouring the gel
- Make 3 mL of 10% ammonium persulfate solution (APS) by dissolving 0.3 g ammonium persulfate and diluting up to 3 mL in ultrapure water.
NOTE: 1.2 mL of APS is needed per gel. Date the tube after making it. APS solution expires in 2 weeks, and should be kept at 4 °C. - Transfer 125 µL of tetramethylethylenediamine (TEMED) to a separate microcentrifuge tube.
NOTE: TEMED is toxic; wear PPE such as gloves, eyeglasses and lab coat whenever handling. - Acquire a squirt bottle and remove the pointed funnel. Measure out 125 mL of water in a graduated cylinder and add to the bottle. Mark the water level on the outside of the bottle with a permanent marker. Discard the water. This modified squirt bottle can be reused indefinitely with proper cleaning (see below).
- Set up the corked rings next to the sink so there is a place to put the gel once it is poured. Place the plate sandwich here. Put a cork in the sink so there is a place to sit the plates on while the gel is being poured. The gel will be poured into the glass plate sandwich at an angle, so the bottom will rest on the cork while the top will rest on the edge of the sink. Make sure there are pads underneath these areas in case polyacrylamide pours off.
- Place APS solution and TEMED solution on the other side of the sink, with the empty squirt bottle. Place the well comb with the desired number of wells on the side of the sink with the plates. Put 4 clamps nearby.
NOTE: The next step in the procedure is very time sensitive. Be prepared to move quickly through these steps. There is a limited amount of time to get the mixture between the plates before it polymerizes. All components (micropipettes preset to correct volume, solutions, glass plates) are carefully arranged to pour the gel as quickly as possible. If a slower polymerization is desired, APS and TEMED concentrations can be halved; this will, however, require a longer polymerization time. - Pour the 20% acrylamide gel solution into the squirt bottle to the marked point. Pour a little more gel solution above the marked line, so there is enough solution in the case of small leaks. This is approximately 120 mL of gel solution.
- Add 120 µL of TEMED and 600 µL of APS to the polyacrylamide gel solution in the squirt bottle at the same time. Quickly add an additional 600 µL of APS to the polyacrylamide gel solution.
- Swiftly cap the squirt bottle and swirl. Place the plate sandwich in the sink.
- Begin pouring. This should only take 1-2 min in order to avoid polymerization. Slowly, but steadily, add pressure to the squirt bottle so the gel solution comes out evenly. If the solution starts spattering irregularly from the squirt bottle, release the pressure so the bottle becomes inflated, and resume pouring. Watch to make sure the gel solution is covering all areas, and it is not leaking from any of the sides. To remove air bubbles, change the angle of the plates. The bubbles will pop at the top, and will run faster if the angle is steeper. Continue pouring until the solution has reached the top of the notched area, and is slightly overflowing the notched edge. Remove all air bubbles.
NOTE: If the plate sandwich leaks, or there is not enough gel solution and the solution does not reach the top of the plates, the gel cannot be used. Wait until the gel has polymerized, and clean accordingly. - Add the well comb to the plate sandwich. Place 4 clamps on the comb to press down and allow for even distribution of the wells.
- Quickly dispose of the remaining gel solution in the squirt bottle into labeled polyacrylamide waste. Rinse out the squirt bottle with DI water 2 - 3 times, through the squirt nozzle.
NOTE: It is important to perform this cleaning quickly to prevent polyacrylamide from clogging in the squirt bottle. If even a small amount of polymerization occurs within the nozzle, the next gel will be poured too slowly and not successfully. Discard squirt bottle if polymerization occurs.
- Make 3 mL of 10% ammonium persulfate solution (APS) by dissolving 0.3 g ammonium persulfate and diluting up to 3 mL in ultrapure water.
- Clean plates
- Running the acrylamide gel
- While gel is being polymerized, make 1x TBE running buffer. Dissolve 17 g of premixed TBE solid into 1 L of ultrapure water. Shake buffer until all of TBE is dissolved.
- Remove all the binder clips from the plate sandwich.
- Use a razor to cut the tape and remove tape from all the edges of the plate sandwich.
- Clean off all the polymerized gel from the glass surfaces. Use a razor along the edges to scrape off remaining gel. Wipe the plates with paper towels and water to remove as much polyacrylamide residue as possible. Excess acrylamide and/or tape can often burn during gel electrophoresis.
- Remove the well comb by pushing it out evenly on both sides.
- Use a razor to cut off the excess gel along the top of the plate sandwich. Slice along the notched edge of the plate.
- Use a syringe and DI water to rinse the wells and remove debris in the wells. Make sure each well can be filled with water, and is not blocked by excess polymerized gel.
- Place the plate sandwich into the apparatus, so that the longer plate is facing outward, and the notched space is facing inward.
- Add clips to clip the plate sandwich and aluminum plate together with the apparatus. Add to the outside of the plate to promote even running.
- Add TBE buffer to cover the base notches and just enough to cover the wells at the top. TBE buffer can be filtered and reused for approximately 5 gels.
- Warm plates for 20 min by running the gel electrophoresis at 50 W.
- After warming the plates, clean the wells. Use a syringe and the TBE buffer in the baths to squirt the remaining buffer salts from the wells. Do this several times to ensure clean wells. If the wells are not clean, the bands will fray and the image will not be interpretable. Although this can be time consuming, we have found it imperative to obtaining good data.
- To the outermost lane on both sides, pipet 5 µL of 95% formamide with bromophenol blue. This is the same solution as QBO, but with bromophenol blue in place of Orange G. This will provide a visual on where to cut the gel for imaging, and how far down the DNA has run.
- For each sample to be analyzed (generated in section 2.1.1), add 3 µL to a single well of the gel. After loading all samples, wait 1 min for samples to settle.
- Put caps on both the top and bottom plastic baths. Attach the wires to the apparatus. Red indicates the positive electrical charge, so it should be at the bottom so that the DNA runs downwards.
- Attach wires to a power source and set to 50 – 55 W. Run the gel for approximately 3 h or until the bromophenol blue dye front has run at least 2/3 down the gel.
- Imaging the gel
NOTE: This gel is extremely thin and can be difficult to handle. Great caution must be taken to prevent ripping and loss of the entire gel.
NOTE: Depending on the DNA label, it may require using different gel imagers. This protocol uses near-infrared fluorescent dyes and imaging of samples was performed on a gel imager (see Table of Materials). The protocol is specifically written for that imager.- When the bromophenol blue dye front has run at least 2/3 down the gel (~ 28 cm), the gel can be stopped by turning off the power. Remove the gel from the apparatus and place on cork rings.
- Separate the plates with a small wedge plate separator. Place the separator at the inside corner of the notches to pull up. Use caution and do not use excessive force; the notches can break if too much pressure is applied.
- Once suction between the plates are released, carefully lift the top plate and determine which plate the gel is on. If the gel sticks on the bottom plate, pull top plate off and place on cork ring. If the gel is on the top plate being lifted, move slowly and the gel may fall back to the bottom plate. If the gel does not fall, again move slowly and pull the remaining part of the gel from the bottom plate, and place the top plate with the gel on a separate cork ring.
- Once the plates are separated, see where the dye is on the gel and remove excess gel from the top and bottom of the gel. Typically, with our constructs, using either an 18 or 23 nucleotide primer and a 45 nucleotide template, there is no DNA between the bromophenol blue dye front and the bottom of the gel. Cut vertically from the dye to the top of the gel. The spaces between the dye and the edges of the gel also have no DNA. Lastly, cut a couple inches below the well space on the gel. There are no nucleic acids at the very top of the gel.
NOTE: Cutting the gel into the smallest size possible makes it significantly easier to transfer onto the imaging device. If the gel is too large, it is more susceptible to ripping during transfer. While we often trim the gel prior to imaging, we recommend using extreme caution when trimming as relevant data can be lost if care is not taken. To check whether removed portions contain additional data, we will often perform an additional scan of the excised portions of the gel to ensure that no data were lost. - Coat the gel with water to prevent drying out.
- Clean the imager surface. Using task wipers, wipe down the surface with water and ethanol. Add a thin coat of water to the surface where the gel will be placed.
- Transfer the desired portion of gel to the wet surface of the Li-Cor.
- Remove air bubbles by gently pressing them out from the gel, and wiping with a task wiper. Use caution as it is very easy to rip the gel by pushing the air bubbles too hard.
- Image the gel according to the manufacturer's protocols. Analyze the gel using available software for the imager.
- While the gel is being imaged, clean the work space by removing excess acrylamide gel, washing plates, and filtering the TBE buffer to be reused.
Representative Results
A successful polyacrylamide gel analysis of a qualitative characterization of overall activity (described in section 2.1, Figure 1) and of steady-state kinetics (described in the note at conclusion of section 2.1, Figure 2) are shown. An unsuccessful polyacrylamide gel analysis is also shown (Figure 3).
Note that no commercially available ladder or molecular marker is used. Occasionally, we will use a known polymerase-substrate combination to create a molecular marker; however, it is important to note that different modified nucleotides have very different electrophoretic mobilities, making comparison of oligonucleotides of the same length but differing nucleotide structure inappropriate.
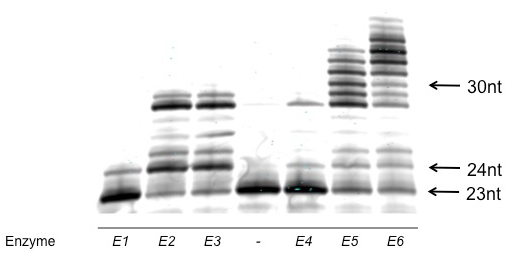
Figure 1: Example of successful polyacrylamide gel analysis of a qualitative characterization of overall activity. Note that the individual bands are well-defined and regular. The bands represent oligonucleotides of differing length; this gel enables easy comparison between polymerases. The no enzyme control lane is indicated with a "-". Activity is judged by both the length of the products and the portion of the labeled primer that is converted to larger products. From this analysis, E6 and E5 are the most active. E3 and E2 are approximately equal in activity, but less than E6 or E5, and E4 and E1 are the least active enzymes.

Figure 2: Example of successful gel for quantitative analysis using steady-state kinetics. Note that the bands are well resolved and well defined. To measure steady-state rate constants of individual steps in oligonucleotide synthesis, an enzyme is incubated with varying quantities of nucleoside triphosphate (in this case, dCTP); note that only the n and (n+1) products are synthesized enabling quantification of the singular event.
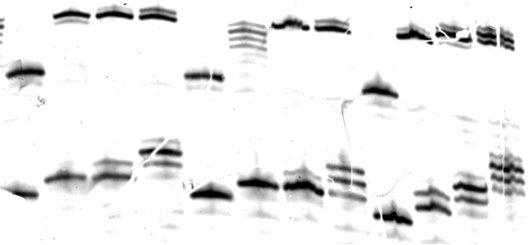
Figure 3: Example of an unsuccessful gel for overall activity. The gel was torn during handling, resulting in visible gaps in bands. Note that the gel bands are irregularly shaped, making quantification and analysis difficult.
Discussion
Here, we have described an assay to characterize the DNA polymerase-mediated synthesis of M-DNA. By using near-infrared labeled DNA primers, and using denaturing polyacrylamide gel electrophoresis to resolve differently sized oligonucleotides, we can obtain single nucleotide resolution on oligonucleotides, enabling precise measurement of synthesis. These approaches can be used to either measure the overall activity of the enzyme (section 2.1) or to measure the Michaelis-Menten parameters of individual steps (note following step 2.1). Our lab has recently used these to both characterize previously evolved enzymes9 as well as rationally engineered enzymes10.
Our assays described here differ from prior methods in their use of a non-radioactive DNA label. Historically, radioactivity has been used to track DNA synthesis due to the high sensitivity that can be obtained using radioactive phosphorous labels11,18. Unfortunately, the high cost of disposal as well as the limited shelf life of radioactive labels can make the use of radioactive labels prohibitive. Here, we describe the use of near-infrared fluorophore labeled DNA, which does not suffer from these drawbacks. Notably, with near-infrared fluorescent dyes we see similar detection limits to radioactively labeled DNA (unpublished results). However, with fluorescent dyes in the visible range, we were not able to observe similar detection limits (unpublished results). While our group typically uses commercially prepared DNA bearing near-infrared fluorophores, these dyes are compatible with a number of established post-synthesis 5' modification chemistries that can be used to install these labels.
Near-infrared fluorescent dyes are also advantageous in that there are multiple commercially available colors, which can enable more complex experiments that monitor synthesis of two labeled oligonucleotides in a single experiment. With radioactive labels, these types of multicomponent experiments are not easily executed. This will likely enable a number of more complex experiments, particularly for orthogonal replication experiments.
This assay, and any assay using denaturing polyacrylamide gel electrophoresis, is primarily limited by the technical challenge of executing the electrophoresis, the low throughput of these experiments, as well as the limited size ranges that are observable at single nucleotide resolution. We hope that this detailed protocol allows groups to overcome the technical challenges of these assays. Notably, while the throughput of the assay is limiting, the use of near-infrared fluorescent labels does increase the throughput, as it does not require the user to develop an autoradiograph. However, the gel still takes approximately 4-6 h to set up and run, limiting the number of experiments that can be run per day. The limited range is caused by the electrophoretic capabilities of polyacrylamide. Practically speaking, these limitations mean that these assays are best for focused research questions that require single nucleotide resolution.
Recently, high throughput DNA sequencing19 has been used increasingly as a method of characterizing DNA polymerases20,21. These assays are noteworthy for their dramatically increased throughput, which enables broader questions focused on sequence biases and error spectra. Importantly, while high throughput sequencing can enable many parallel experiments, it can be challenging to interpret on a single nucleotide basis. This provides an exciting opportunity for enzyme assays employing polyacrylamide gel electrophoresis to fill in the gaps in high throughput sequencing, ensuring that the methods described in this article are relevant for many years to come.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Research Corporation for Scientific Advancement (Cottrell College Scholar Award #22548) and by TriLink Biotechnologies (ResearchReward Grant #G139).
Materials
| Tris HCl | Promega | H5123 | |
| Tris Base | Promega | H5131 | |
| MgCl2 | Fisher Scientific | BP214-500 | |
| Acetylated BSA | Promega | PR-R3961 | |
| KCl | Sigma | P4504 | |
| Dithiothreitol (i.e. DTT) | Research Products International | D11000 | |
| Ethylenediaminetetraacetic acid (i.e. EDTA) (0.5M solution) | Sigma | 03690-100mL | |
| Glycerol | Sigma | G5516 | |
| Formamide | Acros | AC42374-5000 | |
| Orange G | Sigma Aldrich | O3756 | |
| Bromophenol blue | Fisher Scientific | 50-701-6973 | |
| dNTPs | Fisher Scientific | FERR0191 | |
| M-dNTPs (riboNTPs) | Fisher Scientific | 45-001-341 (343, 345, 347) | |
| M-dNTPs (all other modified NTPs) | TriLink Biotechnologies | assorted | |
| primer 1 | IDT DNA / TriLink Biotechnologies | Custom Syntheses | We use the IR700 dye which can be purchased as a custom synthesis. We typically purchase the oligonucleotides HPLC purified. Sequence is 5’-dTAATACGACTCACTATAGGGAGA |
| template 1 | IDT DNA / TriLink Biotechnologies | Custom Syntheses | We typically purchase the oligonucleotides HPLC purified. Sequence is 5’-dCGCTAGGACGGCATTGGATCAGTCTCCCTATAGTGAGTCGTATTA |
| Acrylamide | Research Products International | A11405 | 38.67% acrylamide and 1.33% bis-acrylamide |
| Tris/Borate/EDTA (TBE) solid | Research Products International | T22020 | |
| Urea ultrapure | Research Products International | U20200 | |
| Gel tape | CBS Scientific | GT-72-10 | |
| Large white spring clamp polypropylene | CBS Scientific | GPC-0001 | |
| Ammonium persulfate (APS) | Fisher Scientific | BP179 | |
| Tetramethylethylenediamine (TEMED) | Fisher Scientific | BP15020 | |
| 0.75 mm spacers | CBS Scientific | SGS-20-0740A | |
| 33×42 Notched Glass Plate Set | CBS Scientific | SGP33-040A | |
| Wedge plate separator | CBS Scientific | WPS-100 | |
| Comb for gel electrophoresis | CBS Scientific | SG33-0734 | |
| Gel electrophoresis rig | CBS Scientific | SG-400-33 | |
| ultrapure water | we use a Milli-Q system from Millipore | ||
| DNA polymerases | we prepare these in our laboratory using published protocols. |
Referências
- Ong, J. L., Loakes, D., Jaroslawski, S., Too, K., Holliger, P. Directed evolution of DNA polymerase, RNA polymerase and reverse transcriptase activity in a single polypeptide. J Mol Biol. 361 (3), 537-550 (2006).
- Chen, T., Romesberg, F. E. Directed polymerase evolution. FEBS letters. 588 (2), 219-229 (2014).
- Leconte, A. M., et al. Directed evolution of DNA polymerases for next-generation sequencing. Angew Chem Int Ed Engl. 49 (34), 5921-5924 (2010).
- Xia, G., et al. Directed evolution of novel polymerase activities: mutation of a DNA polymerase into an efficient RNA polymerase. Proc Natl Acad Sci U S A. 99 (10), 6597-6602 (2002).
- Chen, T., et al. Evolution of thermophilic DNA polymerases for the recognition and amplification of C2′-modified DNA. Nat Chem. 8 (6), 556-562 (2016).
- Thirunavukarasu, D., Chen, T., Liu, Z., Hongdilokkul, N., Romesberg, F. E. Selection of 2′-Fluoro-Modified Aptamers with Optimized Properties. J Am Chem Soc. 139 (8), 2892-2895 (2017).
- Taylor, A. I., et al. Catalysts from synthetic genetic polymers. Nature. 518 (7539), 427-430 (2015).
- Alves Ferreira-Bravo, I., Cozens, C., Holliger, P., DeStefano, J. J. Selection of 2′-deoxy-2′-fluoroarabinonucleotide (FANA) aptamers that bind HIV-1 reverse transcriptase with picomolar affinity. Nucleic Acids Res. 43 (20), 9587-9599 (2015).
- Schultz, H. J., et al. Taq DNA Polymerase Mutants and 2′-Modified Sugar Recognition. Bioquímica. 54 (38), 5999-6008 (2015).
- Rosenblum, S. L., et al. Design and discovery of new combinations of mutant DNA polymerases and modified DNA substrates. Chembiochem. 18 (8), 816-823 (2017).
- Creighton, S., Goodman, M. F. Gel kinetic analysis of DNA polymerase fidelity in the presence of proofreading using bacteriophage T4 DNA polymerase. J Biol Chem. 270 (9), 4759-4774 (1995).
- Joyce, C. M., Benkovic, S. J. DNA polymerase fidelity: kinetics, structure, and checkpoints. Bioquímica. 43 (45), 14317-14324 (2004).
- Leconte, A. M., et al. Discovery, characterization, and optimization of an unnatural base pair for expansion of the genetic alphabet. J Am Chem Soc. 130 (7), 2336-2343 (2008).
- Sanger, F., Nicklen, S., Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 74 (12), 5463-5467 (1977).
- Karger, B. L., Guttman, A. DNA Sequencing by Capillary Electrophoresis. Electrophoresis. 30 (Suppl 1), S196-S202 (2009).
- Lawyer, F. C., et al. High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5′ to 3′ exonuclease activity. PCR Methods Appl. 2 (4), 275-287 (1993).
- Lawyer, F. C., et al. Isolation, characterization, and expression in Escherichia coli of the DNA polymerase gene from Thermus aquaticus. J Biol Chem. 264 (11), 6427-6437 (1989).
- Carroll, S. S., Cowart, M., Benkovic, S. J. A mutant of DNA polymerase I (Klenow fragment) with reduced fidelity. Bioquímica. 30 (3), 804-813 (1991).
- Shendure, J., Ji, H. Next-generation DNA sequencing. Nat Biotechnol. 26 (10), 1135-1145 (2008).
- Larsen, A. C., et al. A general strategy for expanding polymerase function by droplet microfluidics. Nat Commun. 7, 11235 (2016).
- Cozens, C., et al. Enzymatic Synthesis of Nucleic Acids with Defined Regioisomeric 2′-5′ Linkages. Angew Chem Int Ed Engl. 54 (51), 15570-15573 (2015).
