The following sections describe the steps required to perform live imaging followed by single cell tracking of multiple neural populations (Figure 1). All the procedures involving animals described in this protocol must be carried out in accordance with the guidelines of the International Council for Laboratory Animal Science (ICLAS).
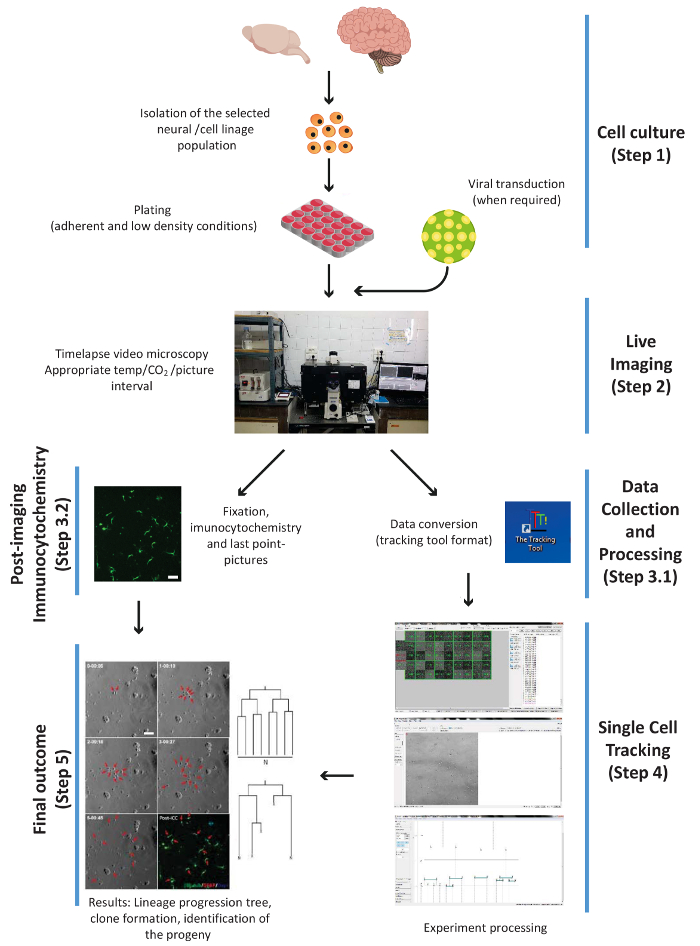
Figure 1. Scheme illustrating the principal experimental steps of the procedure, i.e.: cell culture, live imaging, PICC and data collection, single cell tracking, and the final outcome. The steps are numbered according to the work-flow of the protocol. Please click here to view a larger version of this figure.
1. Cell Culture: Isolation and Plating of the Selected Neural Population or Cell Lineage
NOTE: In conjunction with this protocol, examples of its application to distinct cell populations are given to validate its utility to analyze the biology of neural cells. These include: Adult Neural Stem Cells (aNSCs) derived from the mouse SubEpendymal Zone (SEZ) (for a detailed isolation protocol see14); Postnatal cortical astrocytes to study neuronal reprogramming (for a detailed isolation protocol see15); Postnatal cerebellar astrocytes (for a detailed isolation method see16); and Mouse Neuro-2a Neuroblastoma Cell line (N2a).
- Seed the cells directly on poly-D-lysine coated 24-well plates. Use 1 mL of culture medium per well. Incubate the plates at 37 °C and 8% CO2 for aNSCs, or at 37 °C and 5% CO2 for the astrocytes/cell line for 2 h prior to live imaging. Avoid the use of coverslips to prevent undesired movement as the motorized microscope stage is displaced which makes single cell tracking unfeasible.
NOTE: Cell densities and culture media employed in the experiments are: 30-40,000 cells/well for aNSCs in Dulbecco's modified Eagle's medium (DMEM:F12 Nutrient Mixture medium); 20,000 cells/well for N2a cells in DMEM High Glucose medium, 80,000 cells/well for cerebellar astrocytes in DMEM High Glucose medium; and 55-65,000 cells/well for postnatal astrocytes in DMEM:F12 Nutrient Mixture medium. - Standardize the culture protocol by adjusting the cell density of the culture to the lowest number of cells feasible. Nonetheless, the cell density must be sufficiently high to maintain the viability of the culture.
NOTE: If the cell density is too high, the excess debris or poor dissociation (clumps) may hinder tracking of single cells.
2. Live Imaging by Time-lapse Video-microscopy
- Turn on the microscope, camera, hardware, and incubation systems. Set the temperature and air pressure to 37 °C and 8% CO2 for aNSCs, or to 37 °C and 5% CO2 for the astrocytes/cell line. Allow the temperature and CO2 levels to stabilize for 1-2 h.
NOTE: Specific equipment is required to perform time-lapse video analysis, including: bright field/phase contrast/fluorescence microscopes with motorized components; incubation devices that control the temperature, CO2 and humidity; and finally, reliable and sufficiently powerful hardware and software capable of acquiring and handling the volume of pictures obtained during live imaging experiments (Please check the Table of Materials). - Once the cells are firmly attached to the plate (2 h after plating), use a permanent marker pen to make a small mark on the bottom of one well that will not be used for tracking, i.e., a well that does not contain cells.
NOTE: This mark will be used as a reference to zero the xyz coordinates, and it can be used at any time during or after the experiment, or between the changes of medium, to return to the zero position. - Place the plate inside the microscope's incubation chamber and firmly attach the plate to the stage to avoid any undesired movement during the displacement of the microscope's motorized stage.
- Allow the temperature of the cell culture medium to equilibrate in the chamber for approximately 20 min. This step will avoid a loss of focus during the recording due to the dilation of components.
- Start the live-imaging software and select the time-lapse module to set up the experiment.
- Set the total duration of the experiment and the image acquisition cycles in the "time-schedule tab menu". Due to the inherent phototoxicity of the transmitted or fluorescence light used, define an adequate interval to balance between the temporal resolution of the analysis and the potential cell death.
NOTE: For example, a total of 120 h was selected for aNSC cultures, acquiring brightfield pictures every 5 min. Consider that the acquisition of 120 h of a single movie in this configuration will require 120-150 gigabytes of free storage space in the computer device. - Select the image positions defined by the x and y coordinates, and the focal distance (the z coordinate) in the "xyz points tab menu". Include the reference point (xyz zero coordinate) as the initial position in order to retrieve the coordinates at any time.
- Select the type of acquisition in the "wavelength selection tab menu", brightfield only or in combination with epifluorescence excitation when required. Select the exposure time. Bear in mind that over-exposure to transmitted, and especially fluorescent light, may compromise cell viability (as indicated above).
- For aNSCs, cerebellar astrocytes, and N2a cells, select brightfield (10-50 ms exposure time).
- For transduced cortical astrocytes select brightfield (10-50 ms exposure time) in combination with red/green fluorescence, depending of the reporter used for the experiment (red excitation wavelength: 550 nm and 400 ms exposure time; green excitation wavelength: 460-500 nm and 100 ms exposure time).
- Define the name of the experiment and the folder where the images will be stored. Save the list of positions to reload the experiment at any time, and once all the conditions have been set, run the experiment by clicking on the "run now" button.
- Pause the experiment and re-adjust the focus conditions clicking the "overwrite z button" once per day until the experiment is completed. If changes in the medium are required during the live imaging, pause the experiment and retrieve the plate from the time lapse chamber. Next, change the medium under sterile conditions and place the plate back to the stage (see step 2.3). Re-adjust the focus conditions and resume the experiment.
NOTE: The changes in pH of the medium due to cell death or over-proliferation, as well as variations in room temperature, may affect the correct focusing of the microscope on the cells. For sensitive cultures (such as aNSCs) we recommend the use of medium supplemented with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (final concentration: 1 mM).
3. Post-imaging Immunocytochemistry (PICC), Data Collection, and Processing
- Once the experiment is completed, pause the software and retrieve the plate for fixation and PICC, as described in the next steps.
- Perform cell fixation: wash the cells once with 1 mL phosphate buffered saline (PBS) and add 500 µL of paraformaldehyde (PFA) (4% in PBS), incubating 10 min at room temperature (RT).
CAUTION: Paraformaldehyde is a strong fixative and should be handled carefully to avoid contact with skin or eyes. It must be manipulated only inside a fume hood. - Wash the cells three times with 1 mL of PBS and add 500 µL of the blocking solution (PBS containing 2% (wt/vol) of bovine serum albumin (BSA) and 0.2% (vol/vol) of a non-ionic surfactant). Incubate 1 h at RT.
- Remove the blocking solution and add 250-400 µL of the primary antibodies solution. Incubate 2 h at RT. The primary antibodies solution contains primary antibodies diluted in PBS containing 2% (wt/vol) of BSA and 0.2% (vol/vol) of a non-ionic surfactant. Antibodies used in the experiments described here: GFAP (1:500), βIII-tubulin (1:1,000) and α-tubulin (1:1,000). As this is carried out directly in the well, larger volumes of the solutions are required (250-400 µL) to cover all the cells.
- Wash three times with 1 mL of PBS and add 250-400 µL of the secondary antibodies solution (diluted as described in step 3.4). Secondary antibodies used in the experiments described here: Anti-Mouse Fluorescein (FITC) (1:800), Anti-Rabbit Cy3 (1:500). Incubate 1 h at RT in the dark.
- Wash three times in 1 mL of PBS. Keep the cells in 1 mL of PBS for the subsequent steps of the protocol.
- Place the plate back on the microscope stage and firmly attach it to the stage to avoid undesired movement during the displacement of the motorized microscope stage.
- Retrieve the xyz zero position using the mark made in step 2.2 and re-set the positions to this reference point by pressing the "Offset All X, Y, Z" button. Re-set the focal distance for each position.
- Acquire a final round of images, configuring the conditions necessary for fluorescence emission in the "wavelength selection tab menu" in order to detect the antigens previously targeted in the PICC.
- Briefly, in addition to brightfield, activate FITC (Excitation: 495 nm) and Cy3 (Excitation: 550 nm) acquisition options in the software. Use 10-50 ms for brightfield 400 ms exposure to detect the fluorophores and press the "1 time loop" button, to acquire a final round of pictures.
NOTE: The intensity of the fluorescence may differ depending on the PICC outcome. Adjust the exposition time in order to obtain the optimal image quality.
- Briefly, in addition to brightfield, activate FITC (Excitation: 495 nm) and Cy3 (Excitation: 550 nm) acquisition options in the software. Use 10-50 ms for brightfield 400 ms exposure to detect the fluorophores and press the "1 time loop" button, to acquire a final round of pictures.
- Select the software's file/export option and export the pictures in Tagged Image File Format (Tiff) or Joint Photographic Experts Group format (Jpeg) to a pre-defined destination folder.
- Convert the images exported to the format required by the tracking software: The Tracking Tool17 (tTt). To achieve this, define the input and output folder in the "tTt Converter tool" operating window, as well as the markers used for the positions (xy), channels (c), and time-points (t), and press the "convert images" button.
NOTE: The images must be renamed in accordance with the specific settings and they must be stored in individual folders for each position used in the experiment. Instructions for the installation, requirements, renaming of positions/images, and the use of the tracking tool are available for download at: https://www.bsse.ethz.ch/csd/software/tTt-and-qtfy.html.
4. Single Cell Tracking
- After renaming the data, run the tTt software. Select a user name and tTt work folder.
NOTE: The tracking tool work folder will hold all the analyzed data and the exported results. The work folder must be named tTtexport, containing subfolders named "AVIexport", "Configs", "TreeExport", and "tTtfiles". - Select the experiment to be loaded in the "select experiment folder window", indicating the folder path where the experiment is stored, and then click the "Load experiment" button.
- Run the log file converter to transform the loaded images into a format that can be read by the tracking software (for experiments loaded for the first time, this will be requested automatically by the software).
- Select a position for tracking by clicking on its symbol (after conversion, an overview of the positions recorded during the experiments will be displayed in the "position layout window"). Each position will be represented by a symbol consisting in a picture of the position and its corresponding number (see Figure 1).
- Once the position has been selected and a list of the images available is displayed on the right of the "position layout window", select them and click the "Load images" button.
- Once loading is complete and the "Cell Editor Window" appears, select the wavelengths and image interval to be tracked in the "Cell Editor Window". Wavelength 0 corresponds to brightfield, 1 corresponds to FITC, 2 to Cy3, and 3 to DAPI. In the experiments described here, interval 1 was used, i.e., all images loaded. To clarify, interval 2 means loading of every second image.
- Once the images have been loaded, go back to the "position layout window" and double-click on the icon representing the previously loaded position. The "Movie Window" will appear that allows the single cell tracking to be performed.
- Following the Tracking Tool instructions, proceed to tracking. Select the 0 channel (corresponding to brightfield), and adjust the brightness and contrast ("adjust gamma button"). Start the tracking by pressing the F2 key.
NOTE: During tracking, the tracked cell will be followed by placing the mouse pointer on it and pressing the "0" key. The cell division, cell apoptosis and lost cell buttons are available to monitor these specific cell events. As the experiment is tracked, each of these options will be automatically displayed in the lineage tree drawn in the "cell editor window" as the experiment is being tracked. - Once the clone is tracked, match the brightfield pictures with the immunofluorescence images obtained by PICC to identify the nature of the cell progeny. Each epifluorescence channel will be represented in the "Movie Window" (channel 1, 2, etc.).
5. Final Outcome
- Once single cell tracking has been completed and the progeny identified, save the experiment (Cell Editor Window/File tab/ Save current tree as) and proceed to export the results.
- Export the lineage trees and cell data in the "Export menu" located in the "Cell Editor Window". Likewise, export the cell images and movies via the "Export menu" accessible through the "Movie Window". The images, lineage trees, data, and movies will be exported to the tTt work folder.
The method described enables critical questions regarding the cell biology of multiple neural populations to be resolved. For instance, it has been possible to monitor the progression of the neurogenic and oligodendrogliogenic lineage of aNSCs7,8,14,18. By tracking single aNSCs and their progeny (Figure 2A, B), it was possible to demonstrate that aNSCs isolated in vitro maintain their neurogenic nature, mostly generating neuroblasts, and that they follow a sequence proposed in vivo19 but not previously demonstrated at the single cell level. Moreover, this culture system allowed asymmetric cell divisions to be visualized for the first time in the aNSC lineage from the SEZ (Figure 2B), providing a unique model to study NSC self-renewal8,14. Likewise, and irrespective of the lineage analyzed, it was possible to obtain valuable data regarding cell growth, the rounds of division, cell viability, or cell cycle length.
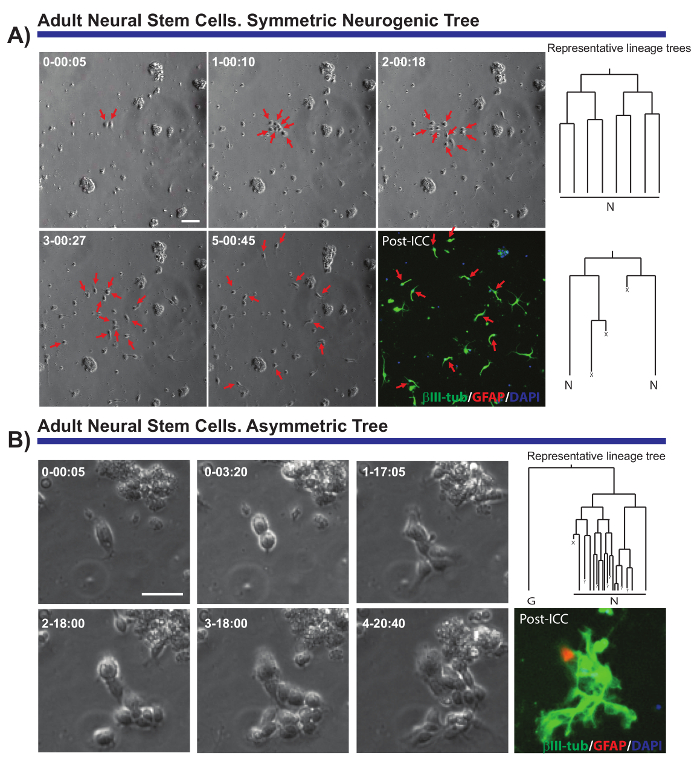
Figure 2. Example of aNSCs isolated from the SEZ, and analyzed by live imaging and single cell tracking. Phase contrast images depict the progression of the clone at different time points (day-h: min). The final image corresponds to the post-imaging immunocytochemistry (PICC) for Glial fibrillary acidic protein (GFAP, red), βIII-tubulin (green) and 4',6-diamidino-2-phenylindole (DAPI, blue). (A) Analysis of symmetric neurogenic trees through different rounds of amplifying divisions to generate post-mitotic neuroblasts. Red arrows point to the cells included in the symmetric trees. On the right, the lineage trees corresponding to the clones and generated by the tTt software are displayed. (B) Example of a progenitor generating an asymmetric neurogenic tree, with one branch undergoing amplifying divisions to produce neuroblasts while the other gives rise to quiescent GFAP positive cells through a potential self-renewal event. On the right, the lineage tree generated by the tTt software is displayed. In all the lineage trees: "N" depicts post-mitotic neuroblasts; "G", quiescent GFAP-positive cells; "X", cell death; and "?" a lost cell. Scale bar represents 50 µm. Please click here to view a larger version of this figure.
Live imaging and single cell tracking analysis also provides an accurate readout of the migratory capacities of a neural population. Such information was obtained from postnatal cerebellar astrocytes submitted to a scratch wound assay20, generating information regarding the average distance traveled by the astrocytes when closing the wound (Figure 3). Moreover, it was possible to see that some of the astrocytes divided during the healing process, while others remain unaltered throughout the experiment. Strikingly, those that divided seem to exhibit more prolific migratory behavior than their non-dividing counterparts (traveling twice as far on average). This phenomenon suggests a very interesting heterogeneity in the astrocytes capacity to form a scar upon injury, which would have been diluted out in the read-out of a classical end-point analysis experiment.
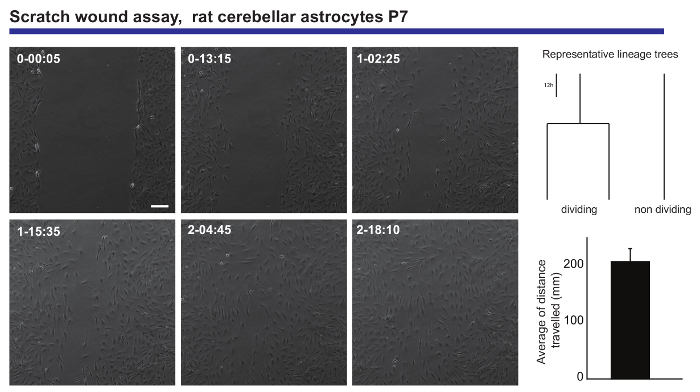
Figure 3. Analysis of the migratory behavior of postnatal cerebellar astrocytes in a scratch wound assay. Phase contrast images depict the wound at different time points (day-h: min). Lineage trees, generated by tTt software, illustrate the representative behavior, in terms of cell division, of the astrocytes while closing the wound. Histogram shows the average distance traveled by the astrocytes analyzed by single cell tracking (mean ± S.E.M.). Scale bar represents 50 µm. Please click here to view a larger version of this figure.
Another interesting feature of the time-lapse video-microscopy experiments is the capacity to compare proliferation and differentiation in a cell population. We tested N2a cells plated under conditions that promote either proliferation (in the presence of 10% fetal bovine serum (FBS)) or differentiation (in the presence of 0.5% FBS + 10 µM arachidonic acid). It was possible to follow the lineage progression of these cells under proliferative conditions (Figure 4A), whereas differentiating cells do not proliferate and they form neurites (Figure 4B). Remarkably, single cell tracking allowed colonies with different proliferation capacities to be distinguished and neurite elongation (and retraction) to be evaluated, providing precise and quantitative data that can subsequently be exported.
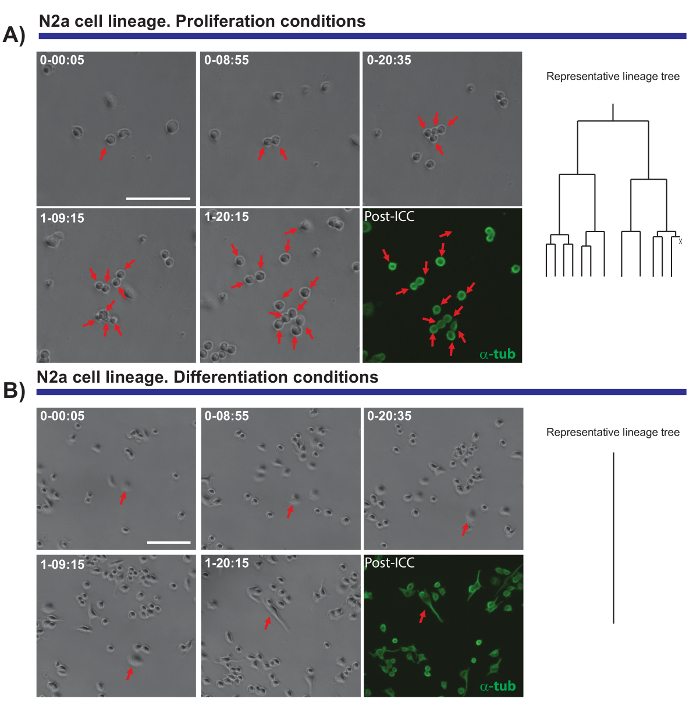
Figure 4. The monitoring of N2a cell biology in proliferation (A) or differentiation conditions (B). Phase contrast images depicting the progression of the clone at different time points (day-h: min). The final image corresponds to the post-imaging immunocytochemistry (PICC) for α-tubulin (green). (A) Single cell tracking allows the rounds of division to be monitored, as well as the heterogeneity in the proliferative response of different cells. On the right, the lineage tree generated by the tTt software illustrates the proliferative behavior of N2a cells. (B) Cells under differentiation conditions exit the cell cycle and generate neurites, a process that can be effectively measured by post-imaging analysis. Single cell tracking, represented by the lineage tree on the right, illustrates how N2a cells exit cell cycle and stop the cell division under differentiation conditions. Scale bar represents 50 µm. Please click here to view a larger version of this figure.
Finally, live imaging and single cell tracking is extremely useful to monitor morphological and molecular changes when cells are submitted to reprogramming. Live imaging of postnatal astrocytes transduced with Achaete-scute homolog 1 (Ascl1) provides valuable data regarding the morphological changes that occur during reprogramming or the blockage of cell division when astrocytes are being reprogrammed (see Figure 5). Moreover, when Ascl1 transduction is combined with the transduction of a construct encoding for Green fluorescence protein (GFP) under the control of the Double Cortin (DCX) promoter, it is possible to define the precise time point when neuronal specific markers begin to be expressed in the reprogrammed cells (Figure 5). Time-lapse video-microscopy also allows the number of cells that successfully complete reprogramming to be quantified and compared to the cells that die during this process. Monitoring such events led to the identification of the critical "checkpoints" in the cells that were successfully reprogrammed9.
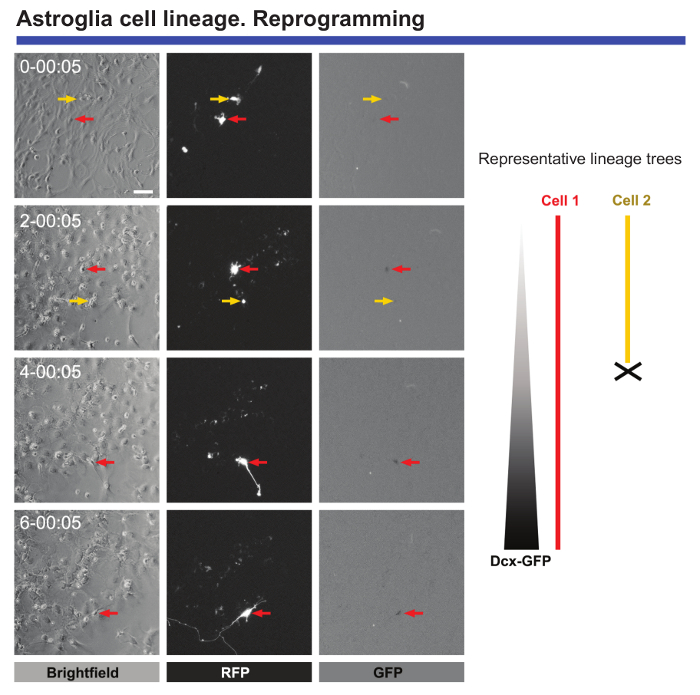
Figure 5. Analysis of postnatal cortical astrocytes subjected to neuronal reprogramming. Reprogramming was induced by transduction with pro-neurogenic Ascl1-Red Fluorescence protein (RFP) vectors. Neuronal conversion was monitored by co-transduction with a vector encoding GFP under the control of a DCX promoter. Phase contrast images show the progression of reprogramming at different time points (day-h: min). Fluorescence images of RFP and GFP expression, respectively. Live imaging and single cell tracking allowed crucial events to be followed, such as morphological changes, the absence of cell division during reprogramming, cell death, and the precise time when reprogrammed cells start to express neuronal markers can be defined. Scale bar represents 80 µm. Please click here to view a larger version of this figure.
